

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 6, стр. 437-455
ТЕРПОЛИМЕРЫ АКРИЛОНИТРИЛА, АКРИЛОВОЙ КИСЛОТЫ И АЛКИЛАКРИЛАТОВ: ВЛИЯНИЕ АЛКИЛАКРИЛАТА НА ТЕРМИЧЕСКИЕ СВОЙСТВА СОПОЛИМЕРОВ
А. Ф. Ващенко a, Р. В. Томс b, М. С. Балашов b, Н. Пичкунов b, А. Ю. Гервальд b, Н. И. Прокопов b, Н. М. Максимов c, А. В. Плуталова a, Е. В. Черникова a, d, *
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
b МИРЭА–Российский технологический университет, Институт тонких химических технологий
им. М.В. Ломоносова
119571 Москва, пр. Вернадского, 86, Россия
c Московский государственный университет имени М.В. Ломоносова. Факультет наук о материалах
119991 Москва, Ленинские горы, 1, стр. 73, Россия
d Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 26.07.2021
После доработки 17.08.2021
Принята к публикации 27.08.2021
Аннотация
Полимеризацией с обратимой передачей цепи под действием несимметричного тритиокарбоната синтезированы терполимеры акрилонитрила с акриловой кислотой и алкилакрилатом; в качестве последнего использовали метил-, бутил-, 2-этилгексил- и лаурилакрилат. Молекулярную структуру терполимеров задавали путем варьирования состава мономерной смеси и способа введения акриловой кислоты и алкилакрилата в реакционную среду – единовременно или непрерывно в ходе синтеза, а молекулярную массу терполимера – концентрацией тритиокарбоната. При исследовании термического поведения синтезированных терполимеров установлено, что температурный интервал циклизации и интенсивность тепловыделения зависят от количества и мольного отношения акриловой кислоты и алкилакрилата. При мольной доле акрилонитрила ~90% природа алкилакрилата существенно не влияет на тепловой эффект циклизации нитрильных групп, однако распределение звеньев акриловой кислоты и акрилонитрила в терполимере влияет на соотношение ионного и радикального механизмов циклизации. Аналогичные эффекты наблюдаются при термообработке терполимеров на воздухе. Анализ изменения химической структуры терполимеров при термообработке в инертной атмосфере и на воздухе показывает, что увеличение длины алкильного заместителя в алкилакрилате благоприятно для повышения длины системы сопряженных связей, а влияние на нее микроструктуры цепи усиливается при переходе от метилакрилата к лаурилакрилату.
ВВЕДЕНИЕ
В последние десятилетия углеродные волокна находят широкое применение для создания полимерных композиционных материалов разного назначения [1–4]. Один из наиболее перспективных источников для получения углеродных волокон – сополимеры акрилонитрила [5, 6]. Наличие относительно дешевого сырья и высокие механические свойства обусловливают возросший интерес к этим сополимерам.
Углеродное волокно на основе ПАН является продуктом многостадийного процесса, включающего стадии синтеза полимера, формования волокна и его термическую обработку. Характеристики конечного продукта в значительной степени зависят от способа и условий проведения каждой стадии. Например, состав сополимеров, распределение сомономеров по цепи, ширина ММР и композиционная неоднородность, которые задаются в ходе синтеза, влияют на свойства сополимеров акрилонитрила (АН) на стадиях формования и термообработки [7–9].
Низкотемпературная стадия термообработки, называемая термостабилизацией, приводит к формированию лестничной структуры полимера, что сопровождается интенсивным выделением тепла [10]. В инертной атмосфере протекает внутри- и межмолекулярная циклизация звеньев ПАН; на воздухе ей сопутствуют реакции окисления и дегидрирования [11]. Эти процессы желательно проводить в широком интервале температур, для чего используют мономеры с карбоксильными или амидными группами, способствующими сдвигу начала циклизации в низкотемпературную область, а также (мет)акрилатные мономеры, которые, напротив, смещают циклизацию в область высоких температур. Первый тип мономеров обеспечивает инициирование циклизации по ионному механизму [12]. Среди карбоновых кислот наибольший эффект достигается благодаря применению итаконовой и акриловой кислот и в меньшей степени метакриловой кислоты [12–15]. (Мет)акрилатные мономеры считаются инертными и не влияют на радикальный механизм циклизации [16].
Среди традиционных промышленных прекурсоров известны как бинарные сополимеры АН с акриловой кислотой (АК), АН с метилакрилатом (МА), так и тройные сополимеры АН с МА и итаконовой кислотой, а также ряд других [13, 16–21]. Однако оптимальный состав ПАН-прекурсоров и его характеристики до сих пор неизвестны, и это один из наиболее дискуссионных вопросов. Тройные сополимеры (терполимеры) удобны возможностью регулировать свойства прекурсора в широких пределах.
В настоящей работе рассматриваются терполимеры на основе АН, АК и алкилакрилатов. Основная часть литературных данных ограничивается терполимерами с метилакрилатом [17, 18, 22, 23]. Алкилакрилаты с более длинным алкильным заместителем представляют не меньший интерес, поскольку дополнительно повышают гибкость цепи и понижают температуру стеклования сополимера, что делает их перспективными в процессах формования.
Ранее мы провели сравнительное исследование бинарных сополимеров АН и АК, а также АН и алкилакрилатов и показали влияние метода синтеза (классическая или контролируемая радикальная полимеризация) и способа введения мономера в синтез (единовременно, непрерывно и полунепрерывно) на реологические и термические свойства сополимеров. Так, при близких средних составах “классических” сополимеров различие в их композиционной неоднородности приводит к непрогнозируемому изменению энергии активации циклизации, степени стабилизации и термостойкости сополимеров. Напротив, для сополимеров того же среднего состава, полученных в условиях обратимой передачей цепи (ОПЦ), изменение микроструктуры цепи, т.е. распределения звеньев акриловой кислоты по цепи, позволяет управлять скоростью циклизации при сохранении высокой термостойкости сополимеров. Увеличение доли АК в сополимере вызывает понижение энергии активации ионной циклизации и повышение энергии активации радикальной циклизации [24]. Рост длины алкилакрилата в бинарных сополимерах, напротив, не влияет на энергию активации радикальной циклизации [25].
В этой связи мы считаем актуальным систематическое исследование влияния длины алкильного заместителя в алкилакрилате на термические свойства терполимеров АН, АК и алкилакрилата. Не исключено, что обнаруженные закономерности позволят расширить спектр сополимеров акрилонитрила, используемых для получения волокон различного назначения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Акрилонитрил (99%), метилакрилат (МА, 97%), н-бутилакрилат (БА, 99%), 2-этилгексилакрилат (ЭГА, 99%), лаурилакрилат (ЛА, 99%) и акриловую кислоту (> 99%) фирмы “Acros” очищали перегонкой по стандартной методике. Лаурилакрилат (“Aldrich”) использовали без дополнительной очистки. ДАК перекристаллизовывали из этанола, хранили в темноте при −3°С. Безводный персульфат калия (ПСК, > 98%) и ОПЦ-агент – 2-циано-2-пропилдодецилтритиокарбонат (CN(CH3)2C–S–C(=S)–S–C12H25, ЦТК, 97%) фирмы “Aldrich” дополнительной очистке не подвергали. ДМСО (99%) и ДМФА (ВЭЖХ) фирмы “Fluka” перед использованием перегоняли.
Для приготовления образцов растворяли ОПЦ- агент в АН из расчета 1 × 10–3 моль/л в конечной реакционной смеси, добавляли АК и алкилакрилат в заданном соотношении и готовили раствор инициатора в ДМСО из расчета 5 × 10–4 или 1 × × 10–3 моль/л в конечной реакционной смеси. Полученные растворы смешивали в объемном соотношении мономеров к растворителю 2 к 3 и готовые смеси разливали по ампулам, подсоединяли их к вакуумной установке и дегазировали путем четырехкратного повторения циклов заморозки–разморозки. По достижении остаточного давления 5 × 10–3 мм рт. ст. ампулы отпаивали.
Полимеризацию проводили при 80°С; по окончании полимеризации ампулы охлаждали до комнатной температуры, вскрывали и при необходимости растворяли полимеризат в ДМСО. Растворы высаживали в деионизованную воду при постоянном перемешивании, выделенный полимер фильтровали и промывали водой. Затем сушили под вакуумом до постоянной массы. Конверсию образцов определяли гравиметрически.
Молекулярно-массовые характеристики сополимеров АН изучали методом ГПХ на хроматографе GPC-120 фирмы “PolymerLabs”. Анализ проводили при 50°С в ДМФА, содержащем 0.1 мас. % LiBr, со скоростью потока 1 мл/мин. Для разделения использовали две колонки PLgel 5 µm MIXED С (М = 5 × 102)–(1 × 107)). Для анализа готовили раствор полимера в элюенте с концентрацией полимера, не превышающей 1 мг/мл и не менее 0.7 мг/мл. ММ рассчитывали по стандартам ПММА и пересчитывали для сополимеров акрилонитрила по известным коэффициентам Марка−Куна−Хаувинка (KПАН = 39.4 × 10–4, α = 0.75, KПMMA = 17.7 × 10–4, α = 0.62 [26]).
Для исследования сополимеров готовили 4%-ный раствор полимеров в ДМСО, наливали его на стеклянную горизонтальную подложку и испаряли растворитель при 80°С до постоянной массы. Готовые пленки снимали с подложки и нарезали квадратные образцы размером 40 × 40 мм; толщина пленки составляла 10–15 мкм.
Тепловые эффекты, наблюдаемые при динамическом нагревании пленок сополимеров, исследовали на дифференциальном сканирующем калориметре “Netzsch DSC 204” фирмы “Netzsch” (Германия) в атмосфере осушенного газа (воздух, аргон) при скорости потока 100 мл/мин в интервале 30–450°С со скоростью нагревания 5–20 град/мин в воздушной атмосфере и 10 град/мин в инертной атмосфере. Для проведения измерений брали приготовленную пленку массой от 4 до 6 мг, помещали в стандартный алюминиевый тигель без крышки. Результаты обрабатывали с помощью программы Netzsch Proteus.
Энергию активации циклизации Ea определяли по методу Киссинджера [27]
(1)
$ - \frac{{{{E}_{a}}}}{R} = \frac{{d\left[ {\ln \left( {\frac{\varphi }{{T_{p}^{2}}}} \right)} \right]}}{{d\left( {\frac{1}{{{{T}_{p}}}}} \right)}},$Состав пленок сополимеров ПАН изучали методом ИК-НПВО-спектроскопии с помощью ИК-фурье спектрометра “Spectrum Two FT-IR Spectrometer” фирмы “Perkin Elmer” в области 4000–400 см–1. В качестве характеристических полос использовали полосы функциональных групп: карбонильной группы акриловой кислоты и сложного эфира (1715–1728 см–1 в зависимости от алкилакрилата) и нитрильной группы акрилонитрила (2229 см–1). Определяли интенсивность характеристических полос поглощения и рассчитывали состав сополимера из калибровочных кривых, построенных из зависимости отношения интенсивности полос поглощения карбонильной группы к нитрильной от мольного отношения акрилового мономера к акрилонитрилу.
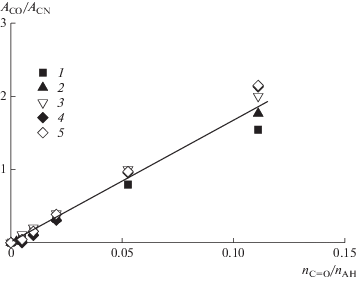
Поскольку разделить в ИК-спектре сигналы, отвечающие карбонильной группе сложного эфира и акриловой кислоты, не представляется возможным, определяли мольное соотношение звеньев АН к акриловым сомономерам (АК и алкилакрилат). В исследуемом интервале составов калибровочные зависимости АН/АК, АН/МА, АН/БА, АН/ЭГА и АН/ЛА близки друг другу (рис. 1), поэтому с достаточной точностью можно определять мольное соотношение звеньев АН к акриловым сомономерам.
Рис. 1.
Начальные участки калибровочных зависимостей смесей АН с АК (1), МА (2), БА (3), ЭГА (4) и ЛА (5) для определения состава сополимера методом ИК-спектроскопии.
Изменения, происходящие в структуре макромолекул при циклизации, изучали методом спектроскопии ИК-НПВО. Для этого пленку образца нагревали при 250°С в аргоне в течение заданного времени, затем регистрировали ИК-спектры при комнатной температуре в режиме НПВО (кристалл алмаз) в области от 4000 до 600 см–1 на ИК-микроскопе “Spectrum Two FT-IR Spectrometer” фирмы “Perkin Elmer” с разрешением 0.5 см–1.
Долю непрореагировавших нитрильных групп φCN [28] и степень стабилизации Es [29] определяли по уравнениям
(2)
${{\varphi }_{{{\text{CN}}}}} = \frac{{{{A}_{{2240\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}{{{{A}_{{2240\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}} + f{{A}_{{1590\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}$(3)
${{E}_{{\text{S}}}} = \frac{{{{A}_{{1590\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}{{{{A}_{{2240\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}$РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез тройных сополимеров
Ранее нами было показано, что АН является более активным мономером в ОПЦ-сополимеризации с алкилакрилатами (МА, БА, ЭГА и ЛА) в ДМСО [25], но менее активным в ОПЦ-сополимеризации с АК [30]. В результате этого в бинарных сополимерах с алкилакрилатами мольная доля АН в сополимере практически не изменяется с ростом конверсии мономеров при содержании АН в мономерной смеси более 80 мол. %, а в сополимерах с АК она понижается при содержании АН в смеси выше 90 мол. %.
Введение в ПАН звеньев АК способствует расширению температурного интервала циклизации и понижению интенсивности тепловыделения за счет реализации дополнительного ионного механизма инициирования циклизации [31]. Алкилакрилаты выполняют функцию внутреннего пластификатора и нарушают регулярность цепи ПАН, смещая процесс циклизации в область высоких температур и также понижая интенсивность тепловыделения. Следовательно, свойства образующихся терполимеров АН можно менять в широких пределах, варьируя состав мономерной смеси.
Для синтеза тройных сополимеров использовали условия, выбранные ранее для бинарных сополимеров АН–АК [24, 30] и АН–алкилакрилат [25], и изменяли соотношение мономеров. Так, вначале при фиксированном мольном отношении АН и АК (98 : 2, 95 : 5, 93 : 7 и 90 : 10%) брали недостаток и эквимолярное отношение АК и алкилакрилата. Предварительные эксперименты показали, что для терполимеров, синтезированных на предельных конверсиях, с увеличением размера алкила в алкилакрилате ширина ММР продуктов реакции повышается от Ð = 1.4–1.5 для МА до Ð = 1.6–1.7 для ЛА. Увеличение доли алкилакрилата по отношению к АК в мономерной смеси способствует увеличению предельной конверсии на 5–10% и, как следствие, небольшому росту молекулярной массы полимера.
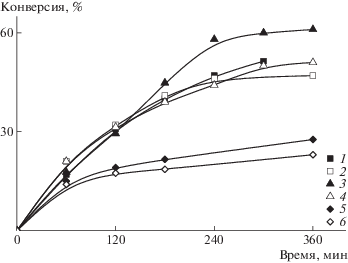
Для доказательства реализации ОПЦ-механизма в исследуемых системах выбрали систему с начальным мольным отношением мономеров в смеси АН : АК = 93 : 7, АК : алкилакрилат = 2 : 1 (89. 8 мол. % АН, 6.8 мол. % АК и 3.4 мол. % алкилакрилата). На рис. 2 приведены зависимости конверсии мономеров от времени при инициировании терполимеризации с помощью ДАК. Видно, что при одинаковом составе мономерной смеси природа алкилакрилата практически не влияет на кинетику сополимеризации за исключением МА (кривая 1).
Рис. 2.
Зависимость конверсии от времени при сополимеризации в системах АН–АК–алкилакрилат МА (1), БА (2), ЭГА (3) и ЛА (4) в ДМСО в присутствии ЦТК и ДАК. Здесь и на рис. 3 и 4, Т = 80°С, массовое содержание мономеров 40%, [ДАК]0 = 5 × × 10–4 моль/л, [ЦТК]0 = 10–3 моль/л, АН : АК : алкил-акрилат = 93 : 7 : 3.5.

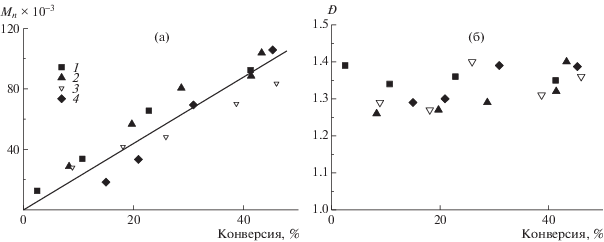
Образующиеся терполимеры характеризуются унимодальным ММР; с ростом конверсии кривые ММР сдвигаются в область высоких ММ (рис. 3). Молекулярная масса терполимеров линейно зависит от конверсии мономеров для всех исследованных систем (рис. 4а). При этом терполимеры характеризуются достаточно узким ММР: дисперсность по ММ составляет 1.2–1.4 (рис. 4б). Наблюдаемые закономерности характерны для ОПЦ-процесса.
Рис. 3.
Нормированные к единичной площади кривые ГПХ терполимеров, образующихся при ОПЦ-сополимеризации в системе АН–АК–алкилакрилат МА (а), БА (б), ЭГА (в) и ЛА (г).
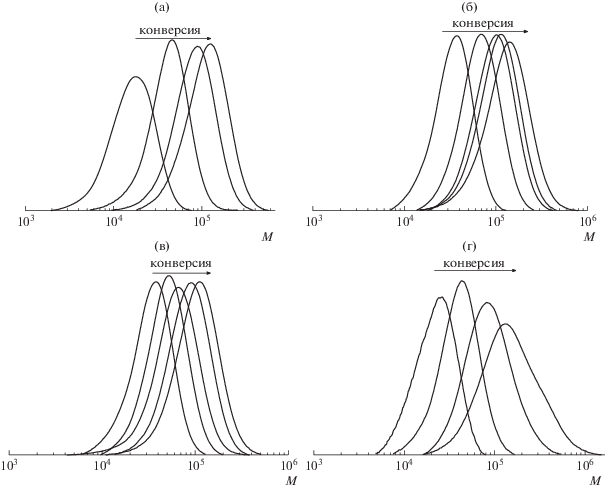
Рис. 4.
Зависимость Mn (а) и дисперсности Ð (б) от конверсии для терполимеров, образующихся при сополимеризации в системе АН–АК–алкилакрилат МА (1), БА (2), ЭГА (3) и ЛА (4).
Ранее мы показали, что изменение способа введения мономеров в реакционную среду (единовременный, полунепрерывный или непрерывный) не влияет на реализацию ОПЦ-механизма [30]. Было проведено две серии экспериментов с использованием мономерной смеси следующего мольного состава АН : АК : алкилакрилат = 98 : 2 : : 10.9 (88.4 мол. % АН, 1.8 мол. % АК и 9.8 мол. % алкилакрилата): в первой мономеры вводили в полимеризацию одновременно, во второй – АК и алкилакрилат непрерывно в ходе полимеризации. Способ введения мономеров не влияет на начальную скорость ОПЦ-сополимеризации, инициированной ПСК, но приводит к небольшому изменению предельной конверсии (рис. 5).
Рис. 5.
Зависимость конверсии от времени в системах АН–АК–алкилакрилат МА (1, 2), БА (3, 4) и ЛА (5, 6) в ДМСО при 55°С, в присутствии [ПСК] = [ЦТК] = 10–3 моль/л. АН : АК : алкилакрилат = 98 : 2 : 10.9; массовое содержание мономеров 40%; единовременное (1, 3, 5) и непрерывное (2, 4, 6) введение АК и алкилакрилата.
Как видно на рис. 6, с ростом конверсии мономеров кривые ГПХ сополимеров сдвигаются в область высоких ММ. Способ введения мономеров в сополимеризацию влияет на ММР конечных продуктов полимеризации: при единовременном способе введения достигаются более высокие конверсии и как следствие более высокая ММ. Одновременно нарастание вязкости приводит к некоторому уширению ММР (табл. 1). Таким образом, как и ожидалось, ОПЦ-механизм реализуется и при изменении способа введения мономеров в синтез.
Рис. 6.
Нормированные к единичной площади кривые ГПХ терполимеров АН–АК–БА, полученных при непрерывном введении АК и БА в сополимеризацию на разных конверсиях под действием ЦТК и ПСК.

Таблица 1.
Характеристики терполимеров АН, АК и алкилакрилат, синтезированных под действием ЦТК из мономерной смеси, содержащей АН : АК : алкилакрилат = 98 : 2 : 10.9
| Алкилакрилат | Способ введения мономеров | Конверсия, % | Mn × 10–3 | Ð |
|---|---|---|---|---|
| МА | Единовременно | 52 | 53.3 | 1.66 |
| Непрерывно | 47 | 49.9 | 1.53 | |
| БА | Единовременно | 61 | 60.6 | 1.70 |
| Непрерывно | 51 | 53.2 | 1.60 | |
| ЛА | Единовременно | 50 | 44.7 | 1.65 |
| Непрерывно | 45 | 38.5 | 1.72 |
Состав терполимеров и его изменение в ходе синтеза изучены методом спектроскопии ИК-НПВО. В ИК-спектрах терполимеров присутствуют характеристические полосы поглощения АН, АК и алкилакрилатов. Валентные колебания протонов основной цепи наблюдаются в области 3000 – 2840 см–1; колебания метиленовых групп СН2 проявляются при 2939 (νsC−H) и 2875 см–1 (νasC−H) и метиновых групп СН при ~2970 см–1. Валентные колебания нитрильной группы АН νCN наблюдаются при 2242 см–1, а карбонильной группы νС=О алкилакрилата – при 1728, 1725, 1726 и 1727 см–1 для МА, БА, ЭГА и ЛА, соответственно. Деформационные колебания δСНН проявляются при 1455 и 1357 см–1, смешанные δСНН и маятниковые ${{\gamma }_{{{\text{C}}{{{\text{H}}}_{2}}}}}$ – при 1057 см–1, слабые полосы поглощения – при 1247 и 762 см–1 [17, 32]. Валентные колебания сложноэфирной группы наблюдаются также при 1170–1150 см–1. В случае МА плечо в области 3000 см–1 относится к валентным колебаниям СН (группы CH3O), а полосы в области 750–960 см–1 относятся к колебаниям метильного и метиленового заместителя сложноэфирной группы. С ростом длины алкильного заместителя повышается интенсивность полос поглощения групп СН2 и происходит переход от общей огибающей к расщеплению на отдельные полосы.
Колебания групп ОН карбоксильной группы и связанной воды проявляются в виде широкой полосы в области 3000–3500 см–1. Полоса, отвечающая карбоксильной группе АК, наблюдается в виде плеча при ~1710 см–1, интенсивность которого возрастает с увеличением доли АК в мономерной смеси.
На рис. 7а видно, что для терполимеров, синтезированных из смеси с мольным отношением компонентов АН : АК : алкилакрилат, равным 93 : 7 : 3.5, и содержащих МА, БА и ЭГА, доля АН в терполимере не изменяется в ходе сополимеризации. В случае терполимера, содержащего ЛА, доля АН в терполимере растет и на конверсии выше 40% сравнивается со значением для терполимеров с другими алкилакрилатами. Для терполимеров с мольным отношением компонентов АН : : АК : алкилакрилат = 98 : 2 : 10.9 доля АН в терполимере постоянна до конверсии ~40%, после чего содержание АН в терполимере постепенно понижается.
Рис. 7.
Конверсионная зависимость состава терполимеров АН–АК–алкилакрилат при мольном отношении мономеров в мономерной смеси АН : АК : алкилакрилат = 93 : 7 : 3.5 (а) и 98 : 2 : 10.9 (б). а: МА (1), БА (2), ЭГА (3) и ЛА (4); б: МА (1), БА (2, 3) и ЛА (4, 5); единовременное (1, 2, 4) и непрерывное (3, 5) введение мономеров.

Таким образом, синтезированные на высоких конверсиях терполимеры характеризуются близким содержанием АН, но разным соотношением АК и алкилакрилата.
Термическое поведение терполимеров акрилонитрила
Реакции циклизации, протекающие в инертной атмосфере. На рис. 8 приведены термограммы сополимеров АН, АК и МА разного состава, зарегистрированные в инертной атмосфере. Видно, что при низком содержании АК в сополимере (АН : : АК = 98 : 2) на термограммах наблюдается один пик, который отвечает протеканию циклизации АН по радикальному механизму.
Рис. 8.
Термограммы сополимеров АН–МА–АК, зарегистрированные в инертной атмосфере при скорости нагревания 10 град/мин. АН : АК = 98 : 2 (а), 95 : 5 (б), 93 : 7 (в) и 90 : 10 (г); АК : МА = 2 : 0 (1), 2 : 2 (2) и 2 : 1 (3) (а); 5 : 0 (1), 5 : 5 (2) и 5 : 1 (3) (б); 7 : 0 (1), 7 : 3.5 (2) и 7 : 1.4 (3); 10 : 0 (1), 10 : 5 (2), 10 : 2 (3) и 10 : 1 (4) (г).
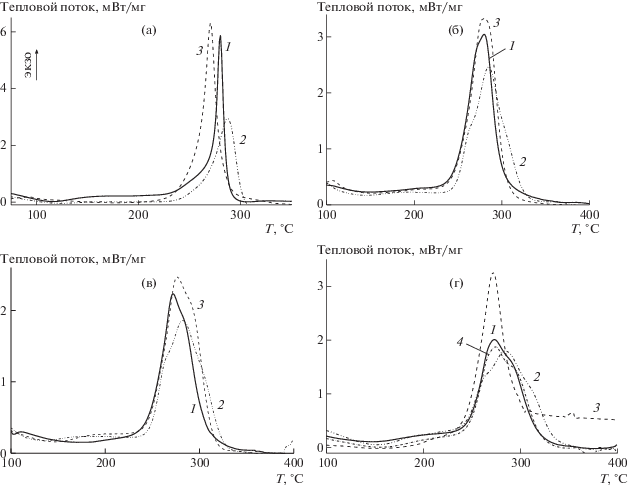

При увеличении доли АК в сополимере (АН : : АК = 93 : 7 и выше) на термограммах появляется второй пик, связанный с низкотемпературной циклизацией АН по ионному механизму:

Звенья алкилакрилата, в данном случае МА, нарушают развитие лестничной структуры вследствие участия в реакции передачи цепи:

В результате совместное введение звеньев МА и АК в ПАН приводит к расширению температурного интервала циклизации; смещению температуры максимума экзо-эффекта в область низких или высоких температур в зависимости от соотношения АК и МА; понижению теплового эффекта при повышении доли МА в терполимере (табл. 2). Аналогичные эффекты наблюдаются при замене МА на другие алкилакрилаты.
Таблица 2.
Анализ термограмм терполимеров АН, АК и алкилакрилата разного состава
| АН : АК : : алкилакрилат | Содержание мономеров в смеси, мол. % | Алкилакрилат | Tпик, °С | –ΔH, Дж/г | ||
|---|---|---|---|---|---|---|
| АН | АК | алкилакрилат | ||||
| 98 : 2 : 2 | 96.0 | 2.0 | 2.0 | МА | 287 | 479 |
| БА | 282 | 596 | ||||
| ЛА | 284 | 566 | ||||
| 98 : 2 : 1 | 97.0 | 2.0 | 1.0 | МА | 270 | 718 |
| БА | 282 | 690 | ||||
| ЛА | 283 | 600 | ||||
| 95 : 5 : 5 | 90.4 | 4.8 | 4.8 | МА | 285 | 589 |
| БА | 261, 292 | 546 | ||||
| ЛА | 262, 291 | 547 | ||||
| 95 : 5 : 1 | 94.0 | 5.0 | 1.0 | МА | 279 | 686 |
| БА | 278 | 766 | ||||
| ЛА | 267, 286 | 686 | ||||
| 93 : 7 : 3.5 | 89.8 | 6.8 | 3.4 | МА | 263, 282 | 490 |
| БА | 261, 289 | 495 | ||||
| ЛА | 263, 279, 293 | 574 | ||||
| 93 : 7 : 1.4 | 91.7 | 6.9 | 1.4 | МА | 276, 290 | 573 |
| БА | 271, 285 | 643 | ||||
| ЛА | 264, 276, 292 | 826 | ||||
| 90 : 10 : 5 | 85.7 | 9.5 | 4.8 | МА | 262, 284 | 578 |
| 90 : 10 : 2 | 88.2 | 9.8 | 2.0 | МА | 272 | 489 |
| 90 : 10 : 1 | 89.0 | 10.0 | 1.0 | МА | 274, 290 | 515 |
Влияние природы алкилакрилата на процесс циклизации в инертной атмосфере исследовали на примере терполимеров, синтезированных на предельной конверсии из мономерной смеси, содержащей 89.8 мол. % АН, 6.8 мол. % АК и 3.4 мол. % алкилакрилата, характеристики которых приведены в табл. 3.
Таблица 3.
Характеристики терполимеров, синтезированных из мономерной смеси, содержащей 89.8 мол. % АН, 6.8 мол. % АК и 3.4 мол. % алкилакрилата
| Образец | Mn × 10–3 | Đ | FАН, мол. % |
|---|---|---|---|
| Т-МА | 75.9 | 1.38 | 91.1 |
| Т-БА | 95.5 | 1.42 | 91.7 |
| Т-ЭГА | 74.9 | 1.40 | 91.9 |
| Т-ЛА | 101.2 | 1.68 | 91.2 |
На рис. 9 представлены термограммы терполимеров в инертной атмосфере. Видно, что природа алкильного заместителя в алкилакрилате практически не влияет на термическое поведение терполимеров. В инертной атмосфере на термограммах наблюдается два пика, связанных с циклизацией по ионному и радикальному механизму. Температура максимума пиков на термограмме, тепловой эффект и его интенсивность практически совпадают для исследуемых образцов за исключением терполимера Т-МА (табл. 4). Температура стеклования терполимеров снижается по мере увеличения длины алкильного заместителя.
Рис. 9.
Термограммы терполимеров. а: Т-МА (1), Т-БА (2), Т-ЭГА (3) и Т-ЛА (4), зарегистрированные в аргоне со скоростью нагревания 10 град/мин; б: Т-МА, зарегистрированные в инертной атмосфере со скоростью нагревания 5 (1), 10 (2), 20 (3) и 30 град/мин (4); в: результаты обработки термограмм образцов Т-МА (1), Т-БА (2), Т-ЭГА (3) и Т-ЛА (4) в координатах уравнения (1).
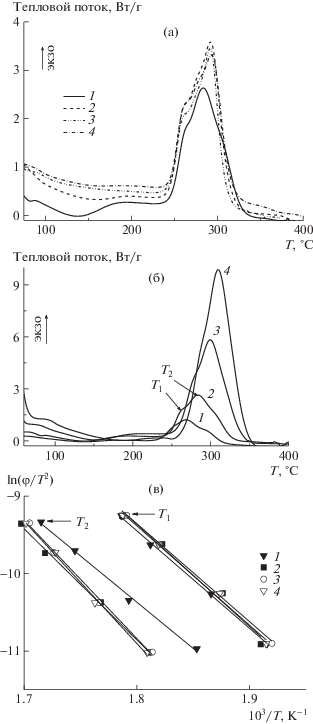
Таблица 4.
Анализ термограмм терполимеров, приведенных на рис. 9
| Терпо-лимер | Тс, °С | Т0, °С | Т1, °С | Tпик, °C | −ΔH, Дж/г | −ΔH/ΔT (Дж/г К) | Ea (циклизации), кДж/моль | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | ионный | ради-кальный | ||||||
| Т-МА | 111 | 233 | 338 | 263 | 285 | 740 | 7.0 | 107 ± 2 | 100 ± 4 |
| Т-БА | 100 | 233 | 333 | 260 | 292 | 890 | 8.9 | 110 ± 10 | 119 ± 4 |
| Т-ЭГА | 100 | 233 | 333 | 260 | 293 | 750 | 7.5 | 105 ± 4 | 127 ± 5 |
| Т-ЛА | 93 | 236 | 333 | 260 | 294 | 800 | 8.2 | 107 ± 5 | 131 ± 5 |
Для оценки энергии активации реакций циклизации использовали подход, предложенный Киссинджером [27]. На рис. 9б для примера приведены термограммы Т-МА, зарегистрированные при разных скоростях нагревания, из анализа которых по уравнению 1 (рис. 9в) рассчитаны значения энергии активации (табл. 4). Видно, что энергия активации ионной циклизации не зависит от природы алкилакрилата и совпадает с точностью до ошибки эксперимента. При учете того, что терполимеры были синтезированы из мономерной смеси одинакового состава и содержание АН в терполимерах близкое, можно полагать, что доля АК в терполимерах также одинаковая. Энергии активации ионной циклизации терполимеров и бинарных сополимеров АН и АК, содержащих ~7 мол. % АК [24], имеют близкие значения. Таким образом, алкилакрилат не оказывает влияния на ионную циклизацию.
Напротив, энергия активации радикальной циклизации в терполимерах зависит от природы алкилакрилата и увеличивается с ростом размера алкильного заместителя в алкилакрилате. В случае бинарных сополимеров АН и алкилакрилата с аналогичным содержанием АН такая зависимость отсутствует [25]. Это кажущееся противоречие, по-видимому, связано с разной микроструктурой цепи полимеров. В случае бинарных сополимеров АН с алкилакрилатами их состав практически не изменяется в ходе сополимеризации. При синтезе терполимеров АК является более активным мономером, чем АН [26], и по грубым оценкам констант сополимеризации по схеме Q–e [26] активность АК в сополимеризации с акрилатами изменяется в ряду МА < БА < ЭГА < ЛА. В свою очередь активность АН в сополимеризации с акрилатами растет в том же ряду [25]. Следовательно, можно ожидать, что терполимеры имеют градиентное строение, и с увеличением длины алкила в алкилакрилате степень градиентности возрастает. Результатом этого должно быть изменение (в данном случае – уменьшение) длины последовательности звеньев АН вдоль цепи, проявляющееся сильнее при переходе от МА к ЛА. К сожалению, это предположение сложно проверить экспериментально из-за относительно небольшого содержания акрилатного мономера в сополимере. Косвенно данное соображение подтверждается тем фактом, что с увеличением размера алкильного заместителя температура максимума пика, соответствующего ионной циклизации, практически не изменяется, а температура максимума пика, отвечающего радикальной циклизации, смещается в область высоких температур.
Если высказанное предположение верно, то изменение микроструктуры цепи повлияет на термическое поведение терполимеров. Действительно, как видно на рис. 10, при единовременном введении мономеров (состав исходной смеси 88.4 мол. % АН, 1.8 мол. % АК и 9.8 мол. % алкилакрилата) интенсивность тепловыделения, связанного с ионной циклизацией, выше, а с радикальной циклизацией – ниже, чем в случае непрерывного введения АК и алкилакрилатов. При этом в первом случае температурный интервал циклизации шире и тепловой поток более равномерный, чем во втором (табл. 5). Изменение способа введения АК и АН приводит к тому, что более активная АК (по сравнению с АН и акрилатами) расходуется более равномерно при непрерывном способе ее введения в синтез, и скорость инициирования ионной циклизации понижается, а вклад радикального механизма усиливается. Непрерывное введение алкилакрилатов, по-видимому, способствует их более равномерному участию в сополимеризации, в результате которого различие между термическим поведением терполимеров с разными алкилакрилатами понижается. Сопоставление данных табл. 4 и 5 показывает, что при примерно равном содержании АН в терполимере расширение температурного интервала циклизации и близкие значения теплового эффекта можно получить, увеличивая долю или АК, или алкилакрилата в сополимере.
Рис. 10.
Термограммы терполимеров, полученных при единовременном (а) и непрерывном (б) способе введения АК и алкилакрилата МА (1), БА (2) и ЛА (3) в сополимеризацию, зарегистрированные в инертной атмосфере, при скорости нагревания 10 град/мин.

Таблица 5.
Анализ термограмм пленок терполимеров АН, АК и алкилакрилата, синтезированных с разным способом введения сомономеров из смеси, содержащей 88.4 мол. % АН, 1.8 мол. % АК и 9.8 мол. % алкилакрилата
| Мономер | Способ введения мономеров | Т0, °С | Т1, °С | Tпик,°С | –ΔH, Дж/г | –ΔH/ΔT, Дж/(г К) | |
|---|---|---|---|---|---|---|---|
| 1 | 2 | ||||||
| МА | Единовременно | 212 | 343 | 252 | 281 | 875 | 6.5 |
| Непрерывно | 226 | 336 | 260 | 286 | 777 | 7.1 | |
| БА | Единовременно | 221 | 364 | 265 | 309 | 738 | 5.2 |
| Непрерывно | 226 | 329 | 262 | 292 | 610 | 5.9 | |
| ЛА | Единовременно | 224 | 311 | 256 | 285 | 625 | 7.2 |
| Непрерывно | 227 | 307 | 251 | 282 | 656 | 8.2 | |
Проследить за химическими превращениями, происходящими в процессе циклизации терполимеров, можно, используя метод ИК-спектроскопии. С учетом данных ДСК пленки терполимеров, синтезированных из смеси с содержанием 89.9 мол. % АН, 6.8 мол. % АК и 3.4 мол. % алкилакрилата, нагревали в течение разного времени при 250°С. Анализ изменений в спектрах позволяет сравнить структурные изменения в макромолекулах с тепловыми эффектами, наблюдаемыми на термограммах. На рис. 11 приведены спектры ИК-НПВО терполимеров Т-МА, Т-БА, Т-ЭГА и Т-ЛА до и после термообработки.
Рис. 11.
ИК-спектры терполимеров Т-МА (а), Т-БА (б), Т-ЭГА (в) и Т-ЛА (г), зарегистрированные после термообработки в инертной атмосфере при 250°С в течение разного времени. Цифры у кривых – время термообработки в минутах.
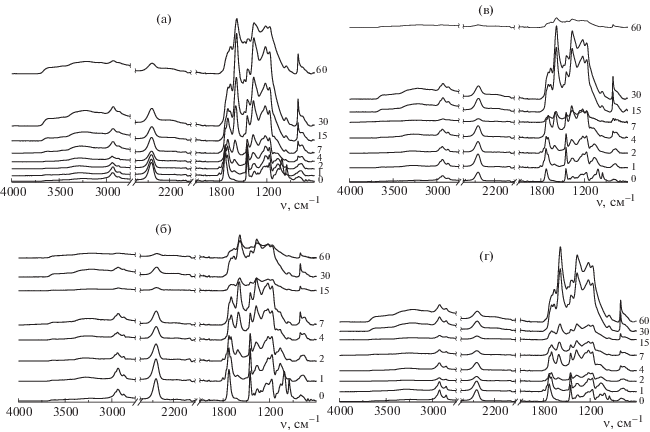
В ходе циклизации нитрильная группа превращается в иминную. Это приводит к понижению интенсивности полосы поглощения 2243 см–1 (νC≡N), относящейся к валентным колебаниям нитрильной группы, и ее уширению в начале и затем к расщеплению на две полосы с максимумами при 2243 см–1 (нитрильная группа в исходном полимере) и при 2200 см–1 (нитрильная группа, участвующая в сопряжении). Одновременно регистрируется полоса поглощения ν−C=N− при 1580–1590 см–1, которая соответствует образованию сопряженной системы за счет протекания внутримолекулярной циклизации. Интенсивность этой полосы повышается по мере термической обработки.
Кроме того, в спектрах появляются новые полосы поглощения при 3350 (νNH), 1376 (δC−H, CH), 1246 и 1172 (νC−N, νC−O), 803 см–1 (δ−C=C−H), которые могут быть отнесены к образующимся в системе сопряженным связям. Полоса поглощения при 1728 см–1, отвечающая карбонильной группе νС=О, при нагревании смещается в длинноволновую область и регистрируется в виде плеча при ~1720 см–1. Одновременно появляется новая полоса при 1660 см–1 в виде плеча полосы валентных колебаний −C=N− (1580 см–1). Согласно литературным данным, эти полосы относят к колебаниям карбонильной группы нафтиридинового и акридонового колец. Их появление связано с инициированием реакции циклизации карбоксильными группами АК и образованием соответствующих циклических структур.
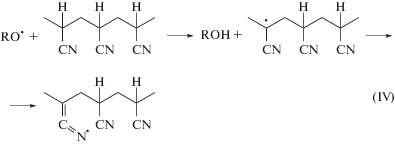
Появление полосы поглощения при 803 см–1, обусловленное образованием кратных связей С=С, является неожиданным, поскольку в инертной атмосфере при 250°С ожидается протекание реакций циклизации. Кратные связи С=С в ПАН формируются обычно при нагревании на воздухе за счет реакций окисления и дегидрирования [11]. Можно предположить, что, наряду с реакцией (IIIб) протекает и реакция (IV), в результате которой могут появиться связи С=С.
Чтобы провести сравнительный количественный анализ скорости превращения нитрильных групп, мы воспользовались уравнениями (2) и (3) и рассчитали долю непрореагировавших нитрильных групп φCN и степень стабилизации Es в ходе циклизации (рис. 12).
Рис. 12.
Зависимости количества остаточных −C≡N-групп φCN (а) и степени стабилизации Es (б) от времени для терполимеров, термически обработанных в инертной атмосфере при 250°С.
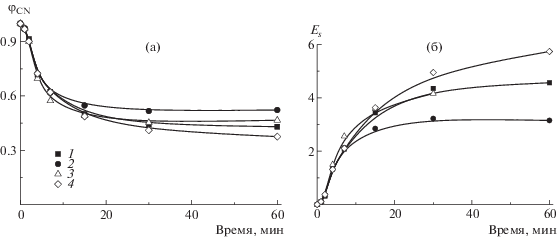
Видно, что в изотермических условиях циклизация развивается достаточно быстро – в течение первых 10 мин нагревания, затем ее скорость снижается, и уже в течение 1 ч доля непрореагировавших групп составляет около 0.4 (рис. 12а). При сравнении с аналогичными данными для бинарных сополимеров [25], можно заметить, что скорость циклизации падает в ряду сополимеров (АН–АК)–(АН–АК–алкилакрилат)–(АН–алкилакрилат). Природа алкилакрилата в терполимерах несущественно влияет на кинетику процесса в отличие от бинарных сополимеров АН–алкилакрилат [25]. По-видимому, это связано с тем, что при данной температуре наиболее вероятна циклизация по ионному механизму за счет звеньев АК. Степень стабилизации Es позволяет оценить развитие системы полисопряженных связей. С увеличением продолжительности термической обработки она повышается для всех исследуемых образцов и максимальна для Т-ЛА. В этом состоит отличие терполимеров от бинарных систем АН–алкилакрилат, для которых характерно замедленное развитие лестничной структуры, наиболее ярко проявляющееся для сополимеров АН с ЛА.
Таким образом, термическое поведение терполимеров можно регулировать, меняя природу алкилакрилата и мольную долю сомономеров. С точки зрения полноты образования системы сопряженных связей наибольший интерес вызывает использование лаурилакрилата.
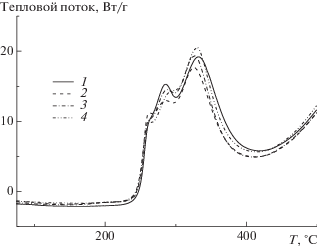
Реакции циклизации, протекающие на воздухе. С точки зрения практического применения более важен процесс термоокислительной стабилизации, протекающий при нагревании на воздухе и включающий реакции циклизации, окисления и дегидрирования. Совокупность этих реакций сопровождается более интенсивным экзо-эффектом, чем циклизация в инертной атмосфере (рис. 13). Если доля АК в терполимере менее 5 мол. %, то увеличение содержания МА приводит к повышению теплового эффекта реакции; при более высоком содержании АК наблюдается обратная ситуация. В целом, чем больше в терполимере звеньев АК и МА, тем меньше интенсивность теплового потока. Аналогичные тенденции поведения терполимеров в воздушной атмосфере проявляются с ростом длины алкильного заместителя в алкилакрилате (табл. 6). При этом с увеличением длины алкильного заместителя в алкилакрилате возрастает интенсивность низкотемпературного пика при АК : алкилакрилат > 2. При дальнейшем повышении содержания алкилакрилата его интенсивность резко понижается, и разница в термическом поведении терполимеров исчезает (рис. 14).
Рис. 13.
Термограммы сополимеров АН–МА–АК, зарегистрированные в атмосфере воздуха, при скорости нагревания 10 град/мин. АН АК = 98 : 2 (а), 95 : 5 (б), 93 : 7 (в) и 90 : 10 (г); АК : МА = 2 : 0 (1), 2.2 (2) и 2 : 1 (3) (а); 5.0 (1), 5 : 5 (2) и 5 : 1 (3) (б); 7 : 0 (1), 7 : 3.5 (2) и 7 : 1.4 (3); 10 : 0 (1), 10 : 5 (2), 10 : 2 (3) и 10 : 1 (4) (г).

Таблица 6.
Анализ термограмм терполимеров АН, АК и алкилакрилата разных составов
| АН : АК : алкилакрилат | Алкилакрилат | Tпик, °С | –ΔH, Дж/г |
|---|---|---|---|
| 98 : 2 : 2 | МА | 276, 313 | 5833 |
| БА | 277, 328 | 4280 | |
| ЛА | 259, 320 | 7759 | |
| 98 : 2 : 1 | МА | 271 | 791 |
| БА | 273, 309 | 6105 | |
| ЛА | 256, 281, 318 | 8154 | |
| 95 : 5 : 5 | МА | 263, 285, 335 | 3684 |
| БА | 262, 288, 336 | 2852 | |
| ЛА | 259, 292, 330 | 7752 | |
| 95 : 5 : 1 | МА | 263, 323 | 2144 |
| БА | 263, 322 | 1752 | |
| ЛА | 260, 309 | 7884 | |
| 93 : 7 : 3.5 | МА | 274, 335 | 3441 |
| БА | 261, 289, 336 | 4359 | |
| ЛА | 265, 338 | 4846 | |
| 93 : 7 : 1.4 | МА | 263, 326 | 4696 |
| БА | 261, 332 | 3452 | |
| ЛА | 266, 341 | 2827 | |
| 90 : 10 : 5 | МА | 280, 349 | 1740 |
| 90 : 10 : 2 | МА | 271, 340 | 493 |
| 90 : 10 : 1 | МА | 267, 339 | 1504 |
Рис. 14.
Термограммы терполимеров Т-МА (1), Т-БА (2), Т-ЭГА (3) и Т-ЛА (4), зарегистрированные на воздухе со скоростью нагревания 10 град/мин.
Изменение микроструктуры цепи должно влиять на термическое поведение терполимеров не только в инертной, но и в воздушной атмосфере. Это видно на рис. 15 при сравнении терполимеров, синтезированных из одинаковой смеси мономеров (88.4 мол. % АН, 1.8 мол. % АК и 9.8 мол. % алкилакрилата), но с использованием разных способов загрузки мономеров: единовременном или непрерывном введении. В первом случае тепловой эффект растет с увеличением длины алкильного заместителя в алкилакрилате, а во втором – в обратном порядке; сами термограммы смещаются в область низких температур при переходе от МА к ЛА (табл. 7).
Рис. 15.
Термограммы терполимеров, полученных при единовременном (а) и непрерывном (б) способе введения АК и алкилакрилата МА (1), БА (2) и ЛА (3) в сополимеризацию, зарегистрированные в атмосфере воздуха, при скорости нагревания 10 град/мин.
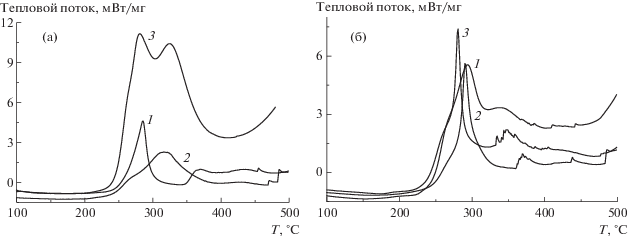
Таблица 7.
Анализ термограмм пленок терполимеров АН, АК и алкилакрилата, синтезированных с разным способом введения сомономеров из смеси, содержащей 88.4 мол. % АН, 1.8 мол. % АК и 9.8 мол. % алкил-акрилата
| Мономер | Способ введения АК и алкилакрилата | Tпик, °С | –ΔH, Дж/г | ||
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| МА | Единовременный | 285 | 366 | – | 807 |
| Непрерывный | 264 | 293 | 336 | 1944 | |
| БА | Единовременный | 268 | 316 | – | 1262 |
| Непрерывный | 266 | 290 | – | 946 | |
| ЛА | Единовременный | 260 | 281 | 324 | 5280 |
| Непрерывный | 258 | 280 | – | 896 | |
Химические процессы, происходящие при нагревании терполимеров на воздухе, исследовали методом ИК-спектроскопии. Пленки нагревали при 225°С, поскольку уже в этих условиях, согласно данным ДСК, регистрируется экзо-эффект. При сравнении ИК-спектров образцов терполимеров, обработанных в инертной атмосфере (рис. 11) и на воздухе (рис. 16) видны небольшие различия, проявляющиеся в области 2850–2920 и 1720–1730 см–1 для терполимеров, содержащих МА, 2850–2920 и 1580–1730 см–1 для терполимеров, содержащих БА, и 1200–1730 см–1 для терполимеров, содержащих ЛА. Эти изменения связаны с дополнительными реакциями окисления и дегидрирования, протекающими на воздухе. Следует заметить, что изменение интенсивностей полос поглощения, связанных с превращением нитрильных групп в иминные, проявляется в спектрах раньше, чем появляется полоса поглощения, отвечающая группе С=С. Таким образом, можно предположить, что реакция циклизации преобладает над реакцией дегидрирования, и реализуется путь, указанный на схеме слева.
Рис. 16.
ИК-спектры терполимеров АН–АК–алкилакрилат МА (а, б), БА (в, г) и ЛА (д, е), синтезированных при единовременной (а, в, д) и непрерывной (б, г, е) загрузке АК и алкилакрилата в реакционную смесь, зарегистрированные после термообработки на воздухе при 225°С в течение разного времени. Цифры у кривых – время термообработки в минутах.
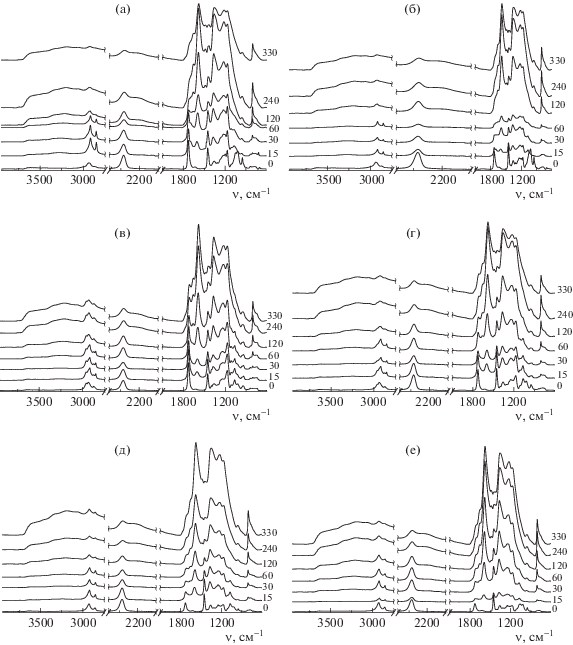

При сравнении спектров терполимеров, полученных при единовременной и непрерывной загрузке АК и алкилакрилата в реакционную смесь видно, что качественные изменения в спектрах не зависят от микроструктуры цепи. Для количественной оценки были рассчитаны доля непрореагировавших нитрильных групп φCN и степень стабилизации Es (рис. 17). В зависимости от алкилакрилата наблюдается разное соотношение скорости расходования нитрильных групп в зависимости от способа синтеза терполимера. Так, для МА конверсия нитрильных групп выше при единовременной загрузке мономеров, конверсия не зависит от способа синтеза для БА и выше при непрерывной загрузке АК и ЛА в реакционную среду (рис. 17а). Степень стабилизации изменяется аналогичным образом и постепенно возрастает с увеличением длины алкильного заместителя. Таким образом, рост длины алкильного заместителя в алкилакрилате благоприятен для повышения длины системы сопряженных связей не только в инертной, но и в воздушной атмосфере. При этом следует отметить, что конверсия нитрильных групп и степень стабилизации выше при обработке терполимера на воздухе, независимо от соотношения в нем кислотных и сложноэфирных групп (рис. 12 и 17).
Рис. 17.
Зависимость степени превращения нитрильных групп φCN (а) и степени стабилизации Es (б) от времени при нагревании на воздухе при 225°С пленок терполимеров АН–АК–алкилакрилат МА (1, 2), БА (3, 4) и ЛА (5, 6), синтезированных при единовременной (1, 3, 5) и непрерывной (2, 4, 6) загрузке АК и алкилакрилата в реакционную смесь.

ЗАКЛЮЧЕНИЕ
Терполимеры акрилонитрила, содержащие мономер – ускоритель циклизации и инертный мономер – пластификатор, представляют интерес для производства углеродных волокон. В настоящей работе для синтеза терполимеров, содержащих АН, АК и алкилакрилат с разной длиной алкильного заместителя, впервые использовали ОПЦ-полимеризацию. Это позволило получить набор полимеров заданной ММ и узким ММР с разным составом и распределением звеньев в цепи.
При термообработке полимеров в инертной атмосфере протекает реакция циклизации нитрильных групп. Вклад ионного и радикального механизмов ее инициирования зависит не только от содержания АК в терполимере, но и от распределения мономерных звеньев в цепи. Акрилатный мономер не влияет на скорость инициирования циклизации по радикальному механизму, но способен нарушать развитие лестничной структуры вследствие участия в реакции передачи цепи. В результате совместное введение звеньев АК и алкилакрилата в ПАН приводит к расширению температурного интервала циклизации и понижению теплового эффекта реакции. При примерно равном содержании АН в терполимере расширение температурного интервала циклизации и близкие значения теплового эффекта можно получить, увеличивая долю или АК, или алкилакрилата в сополимере. Нами обнаружено неожиданное явление – рост энергии активации радикальной циклизации с увеличением длины алкильного заместителя в алкилакрилате, что нехарактерно для бинарных сополимеров АН и алкилакрилатов. Мы предполагаем, что наблюдаемый эффект связан с разным распределением звеньев акрилатного мономера в цепи. Это соображение получило свое подтверждение в независимом эксперименте – изменении микроструктуры цепи при использовании разных способов введения АК и алкилакрилата в синтез. Аналогичные эффекты наблюдаются при термообработке терполимеров на воздухе, в ходе которой протекают экзотермические реакции циклизации, окисления и дегидрирования.
Анализ изменения химической структуры терполимеров при термообработке в инертной атмосфере и на воздухе показывает, что увеличение длины алкильного заместителя в алкилакрилате приводит к повышению степени стабилизации, которая оказывается максимальной для терполимеров, содержащих лаурилакрилат.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-29-17004-мк).
Список литературы
Pakdel E. // Resources, Conservation and Recycling. 2020. 105340.
Yao S.-S., Jin F.-L., Rhee K.Y., Hui D., Park S.-J. // Composites B. 2018. V. 142. P. 241.
Park S.J., Seo M.K. // Polymer Composites. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA. 2012. V. 1.
Zhang J., Chevali V. S., Wang H., Wang C.-H. // Composites B. 2020. 108053.
Rahaman M., Ismail A., Mustafa A. // Polym. Degrad. Stab. 2007. V. 92. № 8. P. 1421.
Frank E., Hermanutz F., Buchmeiser M.R. // Macromol. Mater. Eng. 2012. V. 297. № 6. P. 453.
Tsai J.S., Lin C.H. // J. Appl. Polym. Sci. 1991. V. 42. № 11. P. 3045.
Wilkie C.A., Xue T.J., Mckinney M.A. // Polym. Degrad. Stab. 1997. V. 58. P. 193.
Moskowitz J.D., Wiggins J.S. // Polym. Degrad. Stab. 2016. V. 125. P. 76.
Minagawa M., Okamoto M., Ishizuka O. // J. Polym. Sci., Polym. Chem. Ed. 1978. V. 16. № 11. P. 3031.
Fitzer E., Muller D.J. // Carbon. 1975. V. 13. № 1. P. 63.
Fu Z., Gui Y., Liu S., Wang Z. // J. Appl. Polym. Sci. 2014. V. 131. № 19. 40834.
Bahrami S.H., Bajaj P., Sen K. // J. Appl. Polym. Sci. 2003. V. 88. № 3. P. 685.
Fu Z., Liu B., Deng Y., Ma J., Cao C., Wang J., Ao Y., Zhang H. // J. Appl. Polym. Sci. 2016. V. 133. № 38. P. 43919.
Moskowitz J.D., Wiggins J.S. // Polymer. 2016. V. 84. P. 311.
Alcalá-Sánchez D., Tapia-Picazo J.-C., Bonilla-Petriciolet A., Luna-Bárcenas G., López-Romero J. M., Álvarez-Castillo A. // Int. J. Polym. Sci. 2020. V. 2020. P. 1.
Chernikova E.V., Toms R.V., Prokopov N.I., Duflot V.R., Plutalova A.V., Legkov S.A., Gomzyak V.I. // Polymer Science B. 2017. V. 59 № 1. P. 28.
Chernikova E.V., Kostina Y.V., Efimov M.N., Prokopov N.I., Gerval’d A.Y., Toms R.V. // Polymer Science B. 2015. V. 57. № 2. P. 116.
Jamil S., Daik R., Ahmad I. // Materials. 2014. V. 7. № 9. P. 6207.
Chen H., Pan Y., Hou S., Shao Z., Hong Y., Ju A. // RSC Advances. 2017. V. 7. № 85. P. 54142.
Yılmaz M., Akar A., Köken N., Kızılcan N. // J. Appl. Polym. Sci. 2020. 49023.
Devasia R., Reghunadhan Nair C., Sivadasan P., Ninan K. // Polym. Int. 2005. V. 54. № 8. P. 1110.
Wang J., Zhang M., Fu Z., Zhou T., Xu X., Liu B. // Fibers Polymers. 2015. V. 16. I.12. P. 2505.
Toms R.V., Balashov M.S., Shaova A.A., Gerval’d A.Y., Prokopov N.I., Plutalova A.V., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 2. P. 102.
Toms R.V., Balashov M.S., Gerval’d A.Y., Prokopov N.I., Plutalova A.V., Chernikova E.V. // Polym. Int. 2021.
Polymer Handbook / Ed. by J. Brandrup, E. H. Immergut, E.A. Grulke. New York: Wiley, 1999.
Kissinger H.E. // Anal. Chem. 1957. V. 29. P. 1702.
Collins G.L., Thomas N.W., Williams G.E. // Carbon. 1988. V. 26. № 5. P. 671.
Ouyang Q., Cheng L., Wang H., Li K. // Polym. Degrad. Stab. 2008. V. 93. P. 1415.
Toms R.V., Balashov M.S., Gervald A.Y., Prokopov N.I., Plutalova A.V., Berkovich A.K., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 6. P. 660.
Ismar E., Sarac A.S. // Polym. Adv. Technol. 2016. V. 27. № 10. P. 1383.
Sivy G.T., Coleman M.M. // Carbon. 1981. V. 19. № 2. P. 127.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)