

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 6, стр. 435-449
СИНТЕЗ, ТЕРМИЧЕСКОЕ И ФАЗОВОЕ ПОВЕДЕНИЕ ПОЛИСИЛОКСАНОВ С ПРИВИТЫМИ ДИАЛКИЛЗАМЕЩЕННЫМИ [1]БЕНЗОТИЕНО[3,2-b][1]БЕНЗОТИОФЕНОВЫМИ ГРУППАМИ
Е. А. Заборин a, О. В. Борщев a, М. С. Скоротецкий a, В. В. Городов a, А. В. Бакиров a, М. С. Полинская a, С. Н. Чвалун a, С. А. Пономаренко a, *
a Институт синтетических полимерных материалов им. Н.С. Ениколопова Российской академии наук
117393 Москва, Профсоюзная ул., 70, Россия
* E-mail: ponomarenko@ispm.ru
Поступила в редакцию 04.07.2022
После доработки 13.09.2022
Принята к публикации 20.09.2022
- EDN: AQSOLM
- DOI: 10.31857/S2308113922700164
Аннотация
Синтезирован ряд новых полисилоксанов с привитыми диалкилзамещенными [1]бензотиено[3,2-b][1]бензотиофеновыми группами путем гидросилилирования полиметилсилоксановых матриц с различным соотношением функциональных метилсилоксановых и нефункциональных диметилсилоксановых фрагментов (1 : 5, 1 : 1 и 1 : 0) реакционноспособным 2-ундеценил-7-гексил-[1]бензотиено[3,2-b][1]бензотиофеном. Полученные гребнеобразные полимеры имели среднечисленную молекулярную массу (6–43) × 103 и дисперсностью 1.55–2.79. Исследование термических и термоокислительных свойств показало, что полимер, в котором каждое второе мономерное звено содержит [1]бензотиено[3,2-b][1]бензотиофеновые заместители, имеет наибольшую термоокислительную стабильность (413°С) и наименьший коксовый остаток при 700°С (3%). При этом наименее термостабильным является гребнеобразный полимер с содержанием фрагментов [1]бензотиено[3,2-b][1]бензотиофена 100% и наибольшей молекулярной массой, для которого 5%-ная потеря массы начинается с 274°С. Изучение фазового поведения синтезированных полимеров методом ДСК показало, что увеличение доли фрагментов [1]бензотиено[3,2-b][1]бензотиофена в мономерных звеньях и молекулярной массы способствует повышению температуры фазовых переходов и понижению их энтальпии по сравнению с мономером и силоксановым димером [1]бензотиено[3,2-b][1]бензотиофена. Введение 50 и 100% фрагментов [1]бензотиено[3,2-b][1]бензотиофена в силоксановую полимерную цепь приводит к образованию жидкокристаллических мезофаз с теплотой переходов, характерной для смектиков, что подтверждается результатами поляризационно-микроскопических и рентгенофазовых исследований. Получены значения межслоевого расстояния в смектической мезофазе для полимеров с варьируемым содержанием фрагментов[1]бензотиено[3,2-b][1]бензотиофена и изучено их фазовое поведение. Установлено влияние молекулярной массы на упаковку [1]бензотиено[3,2-b][1]бензотиофена и построены модели взамопроникновения боковых заместителей синтезированных полимеров.
ВВЕДЕНИЕ
Органические и полимерные полупроводники являются актуальным предметом исследований преимущественно благодаря своим уникальным электрическим и оптическим свойствам, сочетающимся с хорошей растворимостью в органических растворителях, гибкостью устройств на их основе и низкой стоимостью технологии их получения [1]. Органические полупроводники применяются в органических полевых транзисторах, органических светодиодах, биосенсорах, органических солнечных батареях и т.д. [2–4]. Исследования органических полупроводников продолжаются как в области синтеза новых материалов, так и в плане совершенствования технологии изготовления различных устройств на их основе.
[1]Бензотиено[3,2-b] [1]бензотиофен (BTBT) и его производные – одни из наиболее эффективных органических полупроводников, используемых в органических полевых транзисторах [5, 6]. Различные производные BTBT находят применение в качестве функциональных материалов в разнообразных устройствах органической электроники: от органических полевых транзисторов [7–9] до фототранзисторов [10], а также в качестве нефуллеренового акцептора в органических солнечных батареях [11, 12]. Исследовано влияние количества атомов углерода (чет-нечетный эффект) в алкил- и алкоксизамещенных BTBT на упаковку надмолекулярных структур, термические и электрические свойства органических полупроводников на их основе [13]. Разницу между физико-химическими свойствами моно- и дизамещенных акилпроизводных BTBT изучали в работах [14, 15]. Органические полевые транзисторы из асимметричных алкил-нафтилпроизводных BTBT демонстрируют хорошие электрические свойства [16]. Тонкие пленки на основе несимметричного децилфенил BTBT показали хорошую растворимость и высокую дырочную подвижность – до 1.3 см2/В с [17].
Однако на сегодняшний день недостаточно внимания уделяется полимерным материалам, содержащим сопряженные фрагменты BTBT. В основном оно сконцентрировано на молекулах, в которых сопряженный фрагмент расположен в основной цепи полимера. По реакции металлорганического синтеза в условиях Сузуки синтезированы сополимеры, содержащие фрагменты BTBT, 9,9-дидецилфлуорена и 4,7-дитиофен-2-ил-бензо(3,1,2)тиадиазола в различном соотношении [18]. Изучение их оптических, электрохимических и фотовольтаических свойств показало, что даже сравнительно небольшое количество фрагментов BTBT в структуре макромолекул приводит к повышению эффективности солнечных батарей на их основе. Органические полевые транзисторы на основе сополимеров BTBT с арилендиимидами продемонстрировали высокую подвижность носителей зарядов для ряда перилендиимидных полимеров, что подтверждает эффективность введения фрагментов BTBT в полимерную цепь с целью повышения эффективности электронных устройств на их основе [19]. Исследования органических полевых транзисторов из сополимера BTBT, тиофена, и дикетопиррола показали, что с увеличением количества фрагментов BTBT в полимерной цепи возрастает дырочная подвижность до 2.47 см2/В с [20]. Там же показано, что увеличение количества BTBT фрагментов существенно не влияет на оптические свойства полученных из таких полимеров пленок и растворов.
Кремнийорганические производные BTBT в настоящее время известны только в виде силоксановых димеров [21]. Это новые материалы для органической электроники, которые совмещают в себе помимо большой дырочной подвижности носителей зарядов, высокую термостабильность, стабильность на воздухе и отличную растворимость в обычных органических растворителях [22]. Уникальность строения таких соединений заключается в сочетании в структуре молекулы фрагментов BTBT, обладающих хорошими полупроводниковыми свойствами, и гибких алкильных спейсеров, значительно улучшающих растворимость. Наличие силоксановых фрагментов позволяет получать самособирающиеся монослойные органические полевые транзисторы, которые находят свое применение в качестве хемосенсоров токсичных газов [23].
В настоящей работе представлены данные о синтезе и свойствах новых полисилоксанов, модифицированных диалкилпроизводными BTBT, химическая формула и схематическая структура которых приведены ниже.

В качестве полимерной матрицы использовали полисилоксаны, содержащие различное количество функциональных гидридсилильных фрагментов, к которым по реакции гидросилилирования присое-диняли 2-ундецил-11-ен-7-гексил[1]бензотиено[3,2-b][1]бензотиофеновые фрагменты. Полученные полимеры исследованы методами термогравиметрического и дифференциально-калориметрического анализа. Их фазовое поведение изучено методом поляризационно-оптической микроскопии.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Анализ методом гель-проникающей хроматографии проводили на хроматографической системе “Shimadzu” с рефрактометром RID10AVP, диодной матрицей SPD-M10AVP, колонки длиной 300 мм и диаметром 7.8 мм (300 × 7.8 мм) (“Phenomenex”, США), заполненной сорбентом the Phenogel (“Phenomenex”, США), размер пор 500 Å, температура термостатирования 40 ± 0.1°С, элюент ТГФ; использовали систему, состоящую из насоса высокого давления СТАЙЕР серия 2 (“Аквилон”, Россия), рефрактометрического детектора “SmartlineRI 2300” (“Knauer”, Германия) и термостата колонок JETSTREAM 2 PLUS ((“Knauer”, Германия). Температура термостатирования 40 ± 0.1°С, элюент ТГФ, скорость потока 1.0 мл/мин, колонки длиной 300 мм и диаметром 7.8 мм (300 × 7.8 мм), заполненной сорбентом Phenogel (“Phenomenex”, США), размер частиц 5 мкм, размер пор 104 Å. Результаты анализа обрабатывали с помощью программы МультиХром 1.6 ГПХ (“Амперсенд”, Россия) с использованием полистирольных стандартов. Для препаративного разделения ГПХ использовали систему “Shimadzu” с рефрактометром RID10A, диодной матрицей SPD-M20A, колонки размером 300 × 21.2 мм) (“Phenomenex”, США), заполненной сорбентом the Phenogel (“Phenomenex”, США), размер пор 104 Å, температура термостатирования 40 ± 0.1°С, элюент ТГФ.
Анализ термических свойств методом дифференциальной сканирующей калориметрии осуществляли на устройстве “Mettler Toledo DSC30” (США) в потоке азота с расходом 50 мл/мин со сканирующей скоростью 20 град/мин в температурном диапазоне –150…+270°С. Термогравиметрический анализ проводили на устройстве “Mettler Toledo TG50” (США) в потоках воздуха и азота с расходом 200 мл/мин.
Мало- и большеугловую дифракцию выполняли на установке “S3-Micropix”, изготовленной фирмой “Hecus” (CuKα-излучение, λ = 1.542 Å). Использовали детектор “Dectris Pilatus 100K” и линейный PSD детектор для больших углов, высокое напряжение на источнике Xenocs Genix трубке и ток составляли 50 кВ и 1 мА соответственно. Для формирования рентгеновского пучка применяли рентгеновскую оптику Fox 3D, диаметр формирующих щелей в коллиматоре Кратки 0.1 и 0.2 мм соответственно. Диапазон измерения углов дифракции 0.05–19.0 нм‑1. Температуру образца в термостатируемой приставке варьировали от комнатной до 300°С с точностью 1°С. Экспозиция составляла 3000 с, калибровку выполняли по стандарту бегената серебра. Спектры ЯМР 1H снимали на спектрометре “Bruker WP-250 SY” (США) на частоте 250 МГц, используя сигнал хлороформа 7.25 м.д. как внутренний стандарт. Фазовое поведение изучали с помощью поляризующего оптического микроскопа “Carl Zeiss Axioscop A40Pol” (Германия).
Методика синтеза исходных полидиметилметилгидросилоксанов (ПМС), содержащих 20% (ПМС20) и 50% (ПМС50) групп Si–H, описана ранее [24]. В качестве полиметилгидросилоксана (ПМС100), содержащего гидридсилильную функцию в каждом звене, использовали коммерчески доступный продукт марки “Пента-804” (Россия). [1]Бензотиено[3,2-b] [1]бензотиен-2-ил)-гексанон (2) получали по методике, опубликованной ранее [16]. 10,11-Дибром-1-(7-гексил [1]бензотиено[3,2-b][1]бензотиен-2-ил)ундеканон (4) синтезировали по реакции ацилирования Фриделя–Крафтса по методике [21].
2-гексил-[1]бензотиено[3,2-b][1]бензотиофен (3) получали по реакции Кижнера–Вольфа. В 1 л двугорлую колбу помещали 1.57 г (4.63 ммоля), соединения 2, 0.94 г (16.7 ммоля) КОН, 3.6 мл гидразин гидрата 80%, 200 мл этиленгликоля. Реакционную смесь кипятили при перемешивании в течение 5 ч, контролируя реакцию методом тонкослойной хроматографии с толуолом в качестве элюента. Реакционную смесь охлаждали до комнатной температуры и экстрагировали из водной фазы диэтиловым эфиром. Органическую фазу концентрировали на роторном испарителе и сушили от остатков органических растворителей на масляном насосе. Полученное соединение очищали методом колоночной хроматографии с циклогексаном в качестве элюента. После очистки и сушки выход продукта составил 1.26 г (84%).
2-гексил-7-ундеценил[1]бензотиено[3,2-b][1]бензотиофен (5) получали восстановлением соединения 4 с использованием LiAlH4 в качестве восстанавливающего агента. В двугорлую колбу объемом 250 мл, содержащую 1 г (1.53 ммоля) соединения 4, приливали 80 мл сухого диэтилового эфира, добавляли 0.9 г (23.7 ммоля) LiAlH4, затем аккуратно вводили 0.4 г (3 ммоля) безводного AlCl3. Реакционную смесь кипятили при перемешивании в течение 40 ч, контролируя степень прохождения реакции методом ТСХ в толуоле. После окончания реакции реакционную смесь аккуратно гасили дистиллированной водой, предварительно охладив колбу и поставив ее в ледяную баню. Затем экстрагировали из водной фазы диэтиловым эфиром. Органическую фазу концентрировали на роторном испарителе и сушили от остатков органических растворителей на масляном насосе. Полученное соединение очищали методом колоночной хроматографии, используя в качестве элюента сначала толуол, а затем циклогексан. После всех этапов очистки выход продукта составил 290 мг (40%).
Синтез полимера Р1. Реакция гидросилилирования
В колбу в инертной атмосфере помещали раствор кремнийорганического полимера ПМС20 (148 мг) в 10 мл сухого толуола, реакционную смесь нагревали до 90°С, затем прикапывали раствор соединения 5 (220 мг, 0.46 ммоля) в 10 мл сухого толуола и 25 мкл катализатора Карстеда. После 13 ч перемешивания при 90°С реакционную смесь пропускали через слой силикагеля в толуоле. Затем смесь концентрировали на роторном испарителе и сушили на масляном насосе. Полученный полимер Р1 очищали на препаративном хроматографе. После очистки и сушки выход продукта составил 102 мг (35%). ЯМР 1H (250 MГц, CDCl3, δ, м.д.): 0.09 (м, 52H), 0.49 (м, 3H), 0.89 (с, 4H), 1.31 (д, 26 H, J = 18.34 Гц), 1.68 (м, 5H), 2.74 (м, 4H), 7.23 (м, 2H), 7.72 (м, 4H).
Синтез полимера Р2
Синтез осуществляли по методике, описанной для полимера Р1 при взаимодействии кремнийорганического полимера ПМС50 (80 мг), соединения 5 (270 мг, 0.56 ммоля) и 25 мкл катализатора Карстеда в 90 мл сухого толуола. Реакцию проводили в течение 3 ч. Полученный полимер Р2 очищали на препаративном ГПХ. После очистки и сушки выход продукта составил 59 мг (15%). ЯМР 1H (250 MГц, CDCl3, δ, м.д.): 0.05 (м, 20H), 0.49 (м, 3H), 0.86 (с, 4H), 1.25 (д, 24H, J = 10.99 Гц), 1.67 (м, 4H), 2.68 (м, 4H), 7.14 (м, 2H), 7.64 (м, 4H).
Синтез полимера Р3
Полимер Р3 синтезировали аналогично полимеру Р1 при взаимодействии кремнийорганического полимера ПМС100 (31 мг), соединения 5 (260 мг, 0.55 ммоля) и 25 мкл катализатора Карстеда в 20 мл сухого толуола в течение 2 ч. Полученный полимер Р3 фракционировали на препаративном ГПХ, после чего выделяли 26 мг Р3a (выход 11%) и 40 мг Р3b (выход 17%). P3a: ЯМР 1H (250 МГц, CDCl3, δ, м.д.): 0.06 (м, 10H), 0.5 (м, 2H), 0.84 (с, 10H), 1.24 (с, 34H), 1.56 (с, 10H), 2.6 (м, 4H), 7.01 (с, 2H), 7.54 (м, 4H). P3b: ЯМР 1H (250 МГц, CDCl3, δ, м.д.): 0.06 (м, 6H), 0.48 (м, 2H), 0.84 (с, 4H), 1.24 (с, 22H), 1.59 (с, 6H), 2.62 (м, 4H), 7.08 (с, 2H), 7.57 (м, 4H).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для выяснения влияния доли привитых фрагментов BTBT на фазовое поведение и свойства целевых макромолекул в работе использовали кремнийорганические сополимеры с содержанием реакционноспособных групп Si–H 20 и 50% (ПМС20 и ПМС50 соответственно). В качестве полимера, содержащего гидридсилильную функцию в каждом звене (100%), был выбран полиметилсилоксан ПМС100.
Основной реакцией для синтеза новых полисилоксанов, модифицированных диалкилпроизводными BTBT, была выбрана реакция гидросилилирования между ПМС и несимметричным диалкилпроизводным BTBT (5), содержащим концевую двойную связь на ундециленовой группе.

Такой выбор был обусловлен доступностью реагентов и возможностью относительно легкого контроля за протеканием реакции методом спектроскопии ЯМР 1Н по исчезновению сигналов протонов от фрагментов –СН=СН2 (характерные мультиплеты при 4.96 и 5.8 м.д.) и Si–H (мультиплет при 4.7 м.д.) (рис. 1).
Рис. 1.
Спектры ЯМР 1Н соединения 5 (а), сополимера ПМС50 (б), реакционной смеси при получении сополимера Р2 (в). Цветные рисунки можно посмотреть в электронной версии.
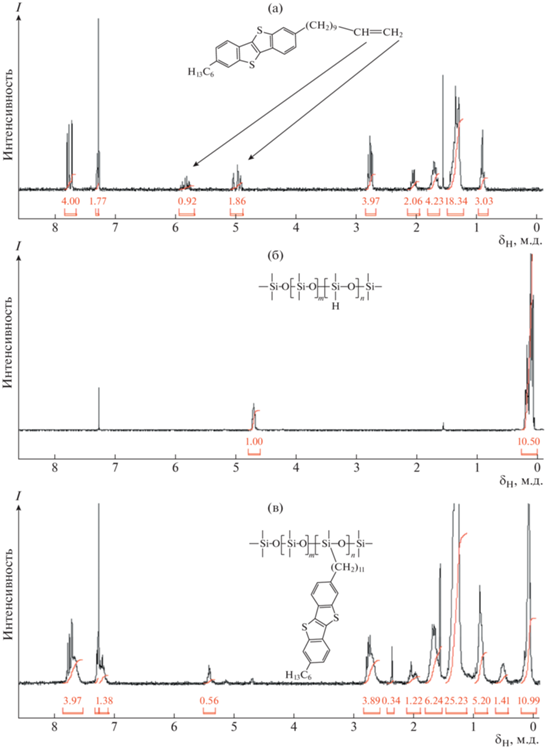
Синтез 2-гексил-(7-ундец-10-ен-1-ил) [1]бензотиено[3,2-b] [1]бензотиофена (5) состоит из четырех стадий, сочетающих в себе последовательное присоединение хлорангидридов в положения 2 и 7 BTBT (1) по методике ацилирования Фриделя–Крафтса и дальнейшее восстановление полученных кетонов.
На первом этапе BTBT ацилировали гексаноил хлоридом по методике [16], получая 1]бензотиено[3,2-b] [1]бензотиен-2-ил)-гексанон (2) с выходом 86%. Это соединение восстанавливали по реакции Кижнера–Вольфа с использованием гидразин гидрата в присутствии гидроксида калия в этиленгликоле до алкильного производного BTBT (3) с выходом 84%. Преимуществами предложенного метода получения соединения 3 по сравнению с описанным ранее методом восстановления соединения 2 боргидридом натрия являются более высокие скорость и выход реакции. Соединение 3 вводили в реакцию ацилирования с 10,11-дибромундеканоил хлоридом в качестве ацилирующего агента. Полученный с выходом 89% кетон 4 восстанавливали LiAlH4 в диэтиловом эфире [25]. Следует отметить, что в последней реакции одновременно с восстановлением кето-группы происходит снятие дибромидной защиты с образованием целевого несимметричного диалкилпроизводного BTBT (5), содержащего концевую двойную связь. Ранее описанный синтез соединения 5 включал последовательное восстановление соединения 3 боргидридом натрия с последующим снятием дибромидной защиты металлическим цинком с суммарным выходом 69% [21]. Попытка восстановить кетон 4 в условиях реакции Кижнера–Вольфа, не увенчалась успехом ввиду образования множества побочных продуктов.
Соединение 5 вводили в реакцию гидросилилирования с различными ПМС в присутствии катализатора Карстеда в безводном толуоле при 90°С. Через 2 ч протекания реакции на спектре ЯМР 1Н пробы, отобранной из реакционной смеси, отсутствовали сигналы протонов концевой двойной связи и сигнал протона при кремнии Si–H, что указывает на полное исчерпание реакционноспособных групп. По данным ГПХ-анализа, в реакционной смеси осталось небольшое количество непрореагировавшего производного BTBT, что обусловлено перегруппировкой кратной связи ундецильного фрагмента из положения 10 углеродной цепи в положение 9, характеризующаяся сигналом при 5.4 м.д. на спектре ЯМР 1Н [26]. В связи с этим все полученные полимеры очищали от побочного продукта методом препаративной ГПХ. При этом полимер Р3 был фракционирован на две фракции – высоко- и низкомолекулярную Р3a и Р3b. Анализ полученных полимеров методом аналитической ГПХ с использованием полистирольных стандартов показал, что их среднечисленные молекулярные массы Мn варьировались в интервале (6.0–43.1) × 103, а дисперсность – от 1.55 до 2.79 (табл. 1).
Таблица 1.
Молекулярно-массовые характеристики полимеров Р1–Р3
| Полимер | Мn × 10–3 | Мw × 10–3 | Дисперсность D |
|---|---|---|---|
| P1 | 12.3 | 33.6 | 2.73 |
| P2 | 21.1 | 42.2 | 2.00 |
| P3a | 43.1 | 120.0 | 2.79 |
| P3b | 6.0 | 9.3 | 1.55 |
По результатам ТГА установлено, что все синтезированные полимеры имеют высокую термическую и термоокислительную стабильность: 5%-ная потеря массы у полимеров Р1, Р2 и Р3b начинается при температурах выше 390°С в атмосфере азота и 325°С на воздухе (табл. 2). При этом наименее термостабильным оказался полимер Р3a с наибольшей молекулярной массой: для него 5%-ная потеря массы начинается с 274°С в обеих средах (рис. 2). Максимальный коксовый остаток (24% при 700°С) в атмосфере азота наблюдается для полимеров Р3а и Р3b с BTBT-заместителями в каждом мономерном звене, т.е. с наибольшим содержанием полиароматических фрагментов. На воздухе величина коксового остатка при 700°С уменьшается в ряду Р1–Р3b–Р3a–Р2 (29, 24, 17 и 3%). Интересно отметить, что полимер P2, в котором каждое второе мономерное звено содержит BTBT-заместители, имеет наибольшую термоокислительную стабильность (413°С) и при этом наименьший коксовый остаток при 700°С (3%).
Таблица 2.
Термические свойства и фазовые характеристики полимеров P1–P3
| Полимер | Потеря 5% массы в потоке газа, °С | Температура фазовых переходов (°С)/энтальпии (Дж/г) | |||
|---|---|---|---|---|---|
| воздух | азот | первое нагревание | охлаждение | второе нагревание | |
| P1 | 348 | 390 | 50/12, 110/5 | – | 52/18, 110/6 |
| P2 | 343 | 413 | 95/24, 156/11 | 153/11, 84/10 | 95/10, 156/11 |
| P3a | 279 | 274 | 90/12, 120/5, 201/10 | 198/10, 109/9 | 118/8, 201/10 |
| P3b | 325 | 404 | 100/14, 122/8, 195/12 | 193/10, 108/11 | 118/11, 193/10 |
| 5 | 296 | 297 | 79/79, 113/16 | 111/15, 52/48 | 70/51, 113/16 |
| O(Si-Und-BTBT-Hex)2 [21] | 316 | 423 | 83/35, 144/18 | 141/17, 76/32 | 81/29, 144/17 |
Рис. 2.
Кривые ТГА полимеров P1 (1), Р2 (2), P3а (3) и P3b (4) при нагревании в атмосфере азота (а) и воздуха (б).

На кривых ДСК полимера Р1 при первом и втором нагревании видны перегибы базовой линии в области –128 и –96°C соответственно, отвечающие за расстекловывание силоксановых фрагментов основной полимерной цепи (рис. 3) [27]. Кроме этого, присутствуют широкие эндотермические пики с максимумами в области 50 и 110°С, с теплотой плавления 12–18 и 6 Дж/г соответственно. Поляризационно-оптическая микроскопия показала, что в скрещенных поляроидах для полимера Р1 при комнатной температуре наблюдается двулучепреломление, интенсивность которого при нагревании постепенно понижается, а выше 120°С полностью исчезает (рис. 4). При этом никаких характерных текстур выявлено не было. Полученные данные свидетельствуют о том, что полимер Р1, имеющий наименьшее содержание жестких ароматических фрагментов BTBT (20%, т.е. в среднем каждое пятое мономерное звено), в области от –96 до +120°С находится в ЖК-состоянии, обусловленном взаимодействием BTBT-фрагментов.
Рис. 3.
Кривые ДСК после первого (а) и второго (б) нагревания полимеров P1 (1), Р2 (2), P3а (3) и P3b (4).

Рис. 4.
Поляризационно-оптические микрофотографии полимеров Р1 (а) и P2 (б) в скрещенных поляроидах при нагревании.

На кривых ДСК полимера Р2 перегибы базовой линии, отвечающей за стеклование полимерной цепи, отсутствуют. При этом проявляются по два экзотермических пика как при первом и втором нагревании, так и при охлаждении. По сравнению с полимером P1 первый экзотермический пик с максимумом при температуре 95°С достаточно узкий, с теплотой 24 и 10 Дж/г при первом и втором нагревании соответственно. Второй экзотермический пик с максимумом при 156°С и теплотой плавления 11 Дж/г воспроизводится как при первом, так и при втором нагревании. При охлаждении видны два эндотермических пика при температурах 153 и 84°С, с теплотой 11 и 10 Дж/г. Таким образом, метод ДСК показал наличие двух обратимых фазовых переходов в области 95 и 156°С с теплотой, характерной для смектических ЖК-мезофаз.
С помощью поляризационно-оптической микроскопии установлено, что в скрещенных поляроидах образца полимера Р2 при комнатной температуре наблюдается двулучепреломление, интенсивность которого повышается в области 156°С и исчезает выше 180°С (рис. 4). При этом никаких характерных текстур выявлено не было. Полученные данные свидетельствуют о том, что для полимера Р2, содержащего 50% мономерных звеньев с BTBT-фрагментами, проявляются две энантиотропные ЖК-мезофазы (предположительно смектические), переходящие выше 180°С в изотропный расплав.
Полимер P3 был разделен на две фракции с различными молекулярными массами: P3а – высокомолекулярный и P3b – низкомолекулярный. При сравнении ДСК-кривых обеих фракций было обнаружено, что они имеют похожее фазовое поведение: на кривых второго нагревания проявляются по два экзотермических пика в области 118 и 201°С (193°С для P3b) с теплотой плавления 8 (11 для P3b) и 10 Дж/г. Оба этих фазовых перехода наблюдаются и на кривых охлаждения, что свидетельствует об их обратимости. На кривых первого нагревания дополнительно проявляются более низкотемпературные широкие эндотермические пики с максимумами при 90 и 100°С, с теплотой 12 и 14 Дж/г для P3а и P3b соответственно. Такая теплота характерна для смектических мезофаз.
На поляризационно-оптических микрофографиях образцов полимера P3а видна шлирен-текстура, которая несколько меняет интенсивность окраски при нагревании (рис. 5). При охлаждении из изотропного расплава при температуре ниже 220°С появляются “батончики”, характерные для смектической мезофазы. Для полимера P3b при нагревании наблюдается разрушенная веерная текcтура, а при охлаждении ниже 205°C появляются “батончики”, которые постепенно превращаются в разрушенную веерную текстуру (рис. 6). Следует отметить, что как шлирен, так и разрушенная веерная текстура характерны для наклонной смектической С мезофазы [28]. Однако без рентгенофазового исследования нельзя однозначно отнести наблюдаемые фазовые переходы к тем или иным мезофазам.
Рис. 5.
Поляризационно-оптические микрофотографии полимера P3a в скрещенных поляроидах при нагревании (а–в) и охлаждении из изотропного расплава (г). Т = 50 (а), 130 (б), 205 (в) и 220°С (г).

Рис. 6.
Поляризационно-оптические микрофотографии полимера P3b в скрещенных поляроидах при нагревании (а–в) и охлаждении из изотропного расплава (г). Т = 50 (а), 110 (б), 150 (в) и 205°С (г).

По результатам анализа кривых ДСК второго нагревания можно выявить зависимость между количеством введенных в полимерную цепь фрагментов BTBT и температурами фазовых переходов полученных сополимеров. У исходного несимметричного диалкилпроизводного BTBT (5) на кривой ДСК два экзотермических пика в области 70 и 113°С с теплотой плавления 51 и 16 Дж/г соответственно (табл. 2) [21]. При переходе к силоксановому димеру O(Si-Und-BTBT-Hex)2, состоящему из двух фрагментов BTBT, соединенных ундецильными спейсерами с центральным тетраметилдисилоксановым фрагментом, фазовое поведение изменяется: температура обоих фазовых переходов повышается (81 и 144°С), при этом теплота плавления первого пика понижается до 29 Дж/г, а второго – немного возрастает (до 17 Дж/г). Такие изменения можно объяснить увеличением массы молекул более чем в два раза при переходе от мономера BTBT (5) к димеру O(Si-Und-BTBT-Hex)2, а также появлением в структуре димера объемного тетраметилдисилоксанового фрагмента, затрудняющего кристаллизацию. При переходе к сополимеру Р2, содержащему 50% мономерных звеньев с BTBT-фрагментами, тенденция роста температуры обоих фазовых переходов сохраняется (95 и 156°С), и происходит незначительное понижение энтальпии до 10 и 11 Дж/г соответственно. При переходе к полимерам Р3 с содержанием BTBT-фрагментов в мономерных звеньях 100% происходит дальнейшее повышение температур обоих фазовых переходов: до 118 и 193°С для низкомолекулярной фракции P3b, а также 118 и 201°С для высокомолекулярной фракции P3a. При этом энтальпии указанных фазовых переходов изменяются несущественно: 11 и 10 Дж/г для P3b, 8 и 10 Дж/г для P3a. Таким образом, в ряду мономер BTBT (5), димер O(Si-Und-BTBT-Hex)2, Р2 и Р3 температура обоих фазовых переходов повышается с ростом количества фрагментов BTBT. Следует отметить, что на температуру первого фазового перехода в основном влияет доля фрагментов BTBT, а на температуру второго – молекулярная масса макромолекулы, что подтверждается при сравнении температур полимеров Р3а и P3b. Введение силоксановых фрагментов в структуру макромолекулы снижает теплоту обоих фазовых переходов в ряду: 5 > O(Si-Und-BTBT-Hex)2 > > P2 ≈ P3a ≈ P3b.
Полимер Р1 выпадает из ряда сравнения ввиду слишком большого содержания силоксановых фрагментов (80%): для него пики обоих фазовых переходов очень широкие (несколько десятков градусов), а их средние температуры существенно ниже, чем у мономера 5. Таким образом, введение 20% групп BTBT несущественно меняет свойства исходного ПМС, что подтверждается проявлением на кривых ДСК низких температур стеклования, характерных для полисилоксанов. Однако при введении 50 и 100% BTBT-фрагментов в мономерные звенья сополимеров P2 и P3 проявляются ярко выраженные пики фазовых переходов, характерные для BTBT-производных [5], возрастают температуры плавления, появляются жидкокристаллические мезофазы с текстурами и теплотами плавления, характерными для смектических мезофаз.
Для определения структуры и фазового поведения синтезированных полимеров был выполнен рентгеноструктурный анализ в малых и больших углах с варьированием температуры образца. Известно, что диалкилзамещенные производные BTBT (Cn-BTBT-Cm) склонны к кристаллизации в решетку моноклинного типа [5], причем ось с направлена под углом к алифатическим и сопряженным фрагментам и приводит к появлению набора 001 рефлексов в малоугловой области. В то же время рефлексы hk0 часто проявляются в большеугловом диапазоне, что свидетельствует об образовании упаковки типа селедочной кости (herring-bone).
Для образца P1 (рис. 7а) при комнатной температуре наблюдались два малоугловых рефлекса 001 и 002, относящиеся к слоевой упаковке. В больших углах также присутствуют три слабо выраженных большеугловых рефлекса при q = 13.3, 15.5 и 18.8 нм–1. Нагревание до 60°C приводило к исчезновению большеугловых рефлексов с одновременным уменьшением полуширины малоугловых пиков, что соответствует переходу в смектическую фазу с увеличенными размерами доменов. Межслоевое расстояние в смектической фазе при этом несколько возрастает от 5.2 до 5.6 нм. Дальнейшее нагревание приводило к переходу в изотропное состояние, а охлаждение восстанавливает исходную фазу.
Рис. 7.
Мало- и большеугловые дифрактограммы для полимеров P1 (а), Р2 (б), P3а (в) и P3b (г) при нагревании от комнатной температуры до 200°C и последующем охлаждении. Интенсивность указана в логарифмическом масштабе, кривые сдвинуты по вертикали.

Дифрактограммы образца P2 (рис. 7б) при комнатной температуре демонстрируют набор рефлексов в малых углах, соответствующих периоду 5.8 нм, также наблюдаются явные большеугловые рефлексы с межплоскостными расстояниями, аналогичными таковым для P1. Нагревание выше 60°C привело к фазовому переходу в смектическую фазу с параметром 4.4 нм с одновременной потерей большеугловых рефлексов. Полученная смектическая фаза переходит в изотропное состояние при температуре выше 140°C и восстанавливается при последующем охлаждении. При этом большеугловые рефлексы восстанавливаются при температуре ниже 80°C. Исходная насцентная кристаллическая фаза не восстанавливается.
Для образцов P3a и P3b (рис. 7в, 7г), являющихся различными фракциями одного полимера, в малоугловой области характерна схожая картина рентгеновского рассеяния: набор широких рефлексов, соответствующих периоду 4.9 нм. Однако для образца P3a с большей молекулярной массой большеугловые рефлексы не наблюдались в отличие от образца P3b. Таким образом, увеличение длины гибкой полимерной цепи препятствует формированию латеральной сопряженной упаковки. Положение большеугловых рефлексов P3b аналогично образцам P1 и P2. Нагревание выше 60°C приводило к необратимому переходу в смектическую фазу с параметром 3.5 нм для обоих образцов, однако в случае образца P3b исчезновение большеугловых рефлексов произошло только выше 100°C, что коррелирует с данными ДСК (двойной пик в области 90–120°C). Обратимый переход в изотропное состояние наблюдался выше 180°C, при этом охлаждение способствовало восстановлению смектической фазы с параметром 3.5 нм, а большеугловые рефлексы в образце P3b появились при температуре ниже 120°C. Насцентная фаза, как и в образце P2, не восстановилась при охлаждении до комнатной температуры.
Таким образом, из данных рентгеноструктурного анализа можно сделать ряд выводов. Во-первых, при увеличении содержания BTBT-фрагментов до 50% и выше возникает насцентная фаза, которая не восстанавливается при последующем охлаждении. Следовательно, для сравнения результатов и анализа фазового поведения более важными являются данные, полученные после охлаждения. Во-вторых, рост молекулярной массы приводит к потере латеральной кристаллической упаковки и взаимодействия сопряженных BTBT-фрагментов. В-третьих, несмотря на сохранение типа BTBT-фрагментов, при увеличении их содержания существенно изменяется межслоевое расстояние, при этом тип упаковки остается неизменным. Предполагаемые модели упаковки представлены на рис. 8, где наглядно проиллюстрировано изменение глубины взаимопроникновения боковых заместителей при повышении их концентрации. Так, для P1, исходя из значений межслоевого расстояния, можно предположить перекрывание только гексильных фрагментов. Для P2 межслоевое расстояние существенно уменьшается, что указывает на сближение ароматических групп от соседних цепей. Для соединений P3a и P3b межслоевое расстояние близко к полной длине заместителя, не учитывая наклона алкильных и сопряженных групп к оси с, что соответствует полному взаимопроникновению боковых фрагментов.
Для образца O(Si-Und-BTBT-Hex)2 значение межслоевого расстояния в конденсированном состоянии составляет 3.58 нм, что весьма близко к упаковке соединений P3a и P3b и является близким к оптимальному для диалкилзамещенных производных BTBT, так, для дизамещенного C11-BTBT это значение составляет 3.59 нм [5].
Рис. 8.
Предполагаемые изменения типа упаковки BTBT-фрагментов для полимеров P1(а), Р2 (б) и P3 (в) в охлажденном состоянии. Указаны параметры c смектической фазы, латеральные параметры приняты a = 5.86, b = 7.74, что является характерными значениями для упаковки диалкилзамещенных BTBT. Заштрихованные области отмечают расположение фрагментов полисилоксана.

ЗАКЛЮЧЕНИЕ
Синтезирован ряд новых полисилоксанов, модифицированных диалкилпроизводными BTBT путем гидросилилирования (со)полисилоксанов с различным содержанием групп Si–H в мономерных звеньях (20, 50 и 100%). При синтезе прекурсора, содержащего BTBT, обнаружено, что в результате восстановления кето-группы LiAlH4 в диэтиловом эфире помимо протекания целевой реакции идет реакция снятия дибромидной защиты с образованием концевой двойной связи, что существенно ускоряет процесс. Химическая структура полученных макромолекул доказана методом ЯМР 1Н, молекулярно-массовое распределение охарактеризовано методом ГПХ. Полученные полимеры имели среднечисленную молекулярную массу (6–43) × 103 и дисперсность 1.55–2.79.
Исследование термических и термоокислительных свойств полученных соединений показало, что полимер Р2, в котором каждое второе мономерное звено содержит BTBT-заместители, имеет наибольшую термоокислительную стабильность (413°C) и наименьший коксовый остаток при 700°С (3%). При этом наименее термостабильным оказался полимер Р3a с содержанием фрагментов BTBT 100% и наибольшей молекулярной массой: для него 5%-ная потеря массы составляет 274°C.
Изучение фазового поведения полученных сополимеров методами ДСК и поляризационной оптической микроскопии дало возможность определить, что полимер Р1 с содержанием жестких фрагментов BTBT 20% в области от –96 до 120°С находится в ЖК-состоянии. Введение 50 и 100% фрагментов BTBT в силоксановую полимерную цепь образует ЖК-мезофазы с теплотой, характерной для смектических мезофаз. Показано, что с увеличением содержания BTBT фрагментов у полимеров Р2, Р3a, P3b повышается температура фазовых переходов. Введение силоксановых фрагментов в структуру макромолекул понижает теплоту обоих фазовых переходов в ряду: мономер BTBT (5) > димер O(Si-Und-BTBT-Hex)2 > > P2 ≈ P3a ≈ P3b.
Данные РСА позволили подтвердить смектическую упаковку соединений, определить зависимость параметра упаковки от содержания BTBT и предложить модели упаковки боковых заместителей. Показано существование насцентных фаз и фазовых переходов, соответствующих потере латерального упорядочения, причем температуры переходов сходны с данными ДСК.
Фазовое поведение синтезированных полимеров с высоким содержанием BTBT-фрагментов указывает на перспективность их использования в устройствах органической электроники, что будет предметом наших дальнейших исследований.
Авторы выражают благодарность Центру коллективного пользования “Центр исследования полимеров ИСПМ РАН” при поддержке Министерства науки и высшего образования Российской Федерации (тема № FFSМ-2021-0005) за проведение ГПХ-анализа и оценки молекулярных масс, а также П.В. Дмитрякову за проведение ТГА и ДСК измерений.
Работа выполнена при финансовой поддержке Российского научного фонда (проект 19-73-30028).
Список литературы
Jiang H., Zhu S., Cui Z., Li Z., Liang Y., Zhu J., Hu P., Zhang H.-L., Hu W. // Chem. Soc. Rev. 2022. V. 51. P. 3071.
Sizov A.S., Agina E.V., Ponomarenko S.A. // Russ. Chem. Rev. 2018. V. 87. № 12. P. 1226.
Shaposhnik P.A., Zapunidi S.A., Shestakov M.V., Agina E.V., Ponomarenko S.A. // Russ. Chem. Rev. 2020. V. 89. № 12. P. 1483.
Trul A.A., Agina E.V., Ponomarenko S.A. // Polymer Science B. 2021. V. 63. № 5. P. 443.
Ebata H., Izawa T., Miyazaki E., Takimiya K., Ikeda M., Kuwabara H., Yui T. // J. Am. Chem. Soc. 2007. V. 129. № 51. P. 15732.
Xie P., Liu T., Sun J., Yang J. // Adv. Funct. Mater. 2022. 2200843.
Li J., Babuji A., Temiño I., Salzillo T., D’Amiso F., Pfattner R., Osal S., Barrena E., Mas-Torrent M. // Adv. Mater. Technol. 2022. 2101535.
Temiño I., Del Pozo F.G., Ajayakumar M.R., Galindo S., Puigdollers J., Mas-Torrent M. // Adv. Mater. Technol. 2016. 1600090.
Shen T., Zhou H., Xin J., Fan Q., Yang Z., Wang J., Mei T., Wang X., Wang N., Li J. // Appl. Surf. Sci. 2019. V. 498. 143822.
Zhang M., Wu J., Lin H., Zhang X., Xu J.-L., Ya Y., Wang S.-D., Wong M., Kwok H.-S. // Electron Device Letters. 2021. V. 42. № 7. P. 998.
Wang Y., Meijia S., Xin K., Xiangjian W., Guangsheng Y. // Polymer. 2019. V. 185. 121976.
Zhijie Z., Jiamin D., Linglong Y., Guo W., Bin Z., Songting T., Ping S., Ryu H.S., Young W.H., Yanming S. // J. Mater. Chem. A. 2020. V. 8. P. 9684.
Roche G.H., Bruckner G., Dumitrescu D.G., Moreau J.J.E., van der Lee A., Wantz G., Dautel O.J. // Adv. Electron. Mater. 2022. V. 8. 2100265.
Christos G., Slaude N., Christian R., Geerts Y.H., Floudas G. // J. Phys. Chem. B. 2014. V. 118. № 5. P. 1443.
Prakoso S.P., Ke Y.-J., Huang D.-S., Wang S.-L., Tao Y.-T. // J. Chin. Chem. Soc. 2022. V. 6 № 3. P. 440.
Dong Y., Sun Y., Liu J., Shi X., Li H., Zhang J., Li S., Yi Y., Mo S., Fan L., Jiang L. // Adv. Sci. 2022. V. 9. 2106085.
Tamayo A., Hofer S., Salzillo T., Ruzié C., Schweisher G., Resel R., Mas-Torrent M. // J. Mater. Chem. S. 2021. V. 9. P. 7186.
Shin W., Jo M.Y., You D.S., Jeong Y.S., Yoon D.Y., Kang J.-W., Sho J.H., Lee G.D., Hong S.-S., Kim J.H. // Synth. Met. 2012. V. 162. № 9–10. P. 768.
Samanta S.K., Song I., Yoo J.H., Oh J.H. // ACS Appl. Mater. Interfaces. 2018. V. 10. № 38. P. 32444.
Nair V.S., Sun Jibin, Qi P., Yang S., Liu Z., Zhang D., Ajayaghosh A. // Macromolecules. 2016. V. 49. № 17. P. 6334.
Borshchev O.V., Sizov A.S., Agina E.V., Bessonov A.A., Ponomarenko S.A. // Chem. Commun. 2017. V. 53. P. 885.
Trul A.A., Chekusova V.P., Anisimov D.S., Borshchev O.V., Polinskaya M.S., Agina E.V., Ponomarenko S.A. // Adv. Electron. Mater. 2022. V. 8. 2101039.
Trul A.A., Sizov A.S., Chekusova V.P., Borshchev O.B., Agina E.V., Shcherbina M.A., Bakirov A.V., Chvalun S.N., Ponomarenko S.A. // J. Mater. Chem. 2018. V. 6. P. 9649.
Gorodov V.V., Demchenko N.V., Buzin M.I. // Russ. Chem. Bull. 2017. V. 66. P. 1290.
Osterod F., Peters L., Kraft A., Sano T., Morrison J.J., Feeder N., Holmes A.B. // J. Mater. Chem. 2001. V. 11. P. 1625.
Ponomarenko S.A., Borshchev O.V., Meyer-Friedrishsen T., Pleshkova A.P., Setayesh S., Smits E.S.P., Mathijssen S.G.J., de Leeuw D.M., Kirchmeyer S., Muzafarov A.M. // Organometallics. 2010. V. 29. № 19. P. 4213.
Klonos P.A. // Polymer. 2018. V. 159. P. 169.
Dierking I. // Textures of Liquid Crystals. Weinheim: Wiley-VSH, 2003.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)