

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 6, стр. 450-458
СИНТЕЗ СМЕШАННЫХ ФУНКЦИОНАЛЬНЫХ ОЛИГОАРИЛОКСИЦИКЛОТРИФОСФАЗЕНОВ
Ю. В. Биличенко a, Фам Ван Тхуан a, Р. С. Борисов a, b, В. В. Киреев a, *
a Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
b Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: kireev.v.v@muctr.ru
Поступила в редакцию 05.10.2022
После доработки 10.11.2022
Принята к публикации 20.11.2022
- EDN: CESLHO
- DOI: 10.31857/S2308113922700267
Аннотация
Олигомерные арилоксициклотрифосфазены со смешанными функциональными группами синтезированы взаимодействием гексахлорциклотрифосфазена и двух фенолов – метил-4-гидроксибензоата (парабен) и 4-аллил-2-метоксифенола (эвгенол) с различной последовательностью их введения. Гидролизом сложноэфирных групп арилоксициклотрифосфазенов получены соответствующие карбокси-феноксициклофосфазены, а окислением аллильных групп м-хлорнадбензойной кислотой – эпоксидные производные. Найдены оптимальные условия указанных превращений строение образующихся олигомеров установлено спектроскопией ЯМР 1Н и ЯМР 31Р, а также методом MALDI-TOF-масс-спектрометрии.
В последние годы синтезированы многочисленные функциональные олигоциклотрифосфазены, содержащие в связанных с атомами фосфора ароматических радикалах различные функциональные группы ‒ гидроксиарилоксидные [1‒6], эпоксидные [7‒9] и другие [10].
Многие из этих соединений находят применение для синтеза полимеров и модификаторов полимерных композиционных материалов. В последнем случае функциональные олигофосфазены используют для улучшения механических и физико-химических характеристик отвержденных полимерных композиционных материалов за счет регулирования параметров образующейся сетки.
Не менее важным является также взаимодействия связующего с поверхностью наполнителя, которые можно изменять природой и числом функциональных групп, влияющих на межфазные процессы в системе наполнитель‒связующее.
Цель настоящей работы – синтез олигомерных арилоксициклотрифосфазенов, содержащих в составе молекул одновременно различные функциональные группы, а также изучение некоторых превращений этих групп.
На схеме представлены исследованные реакции гексахлорциклотрифосфазена и двух исходных фенолов – метилгидроксибензоата (парабен, ParOH) и 4-алилл-2-метоксифенола (эвгенол, EvgOH) с различной последовательностью их введения в реакционную смесь.
Для удобства в схеме и последующем изложении метилпарабен и эвгенол представлены формулами ParOH и EvgOH соответственно (см. схему).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные реагенты
Гексахлорциклотрифосфазен – белое кристаллическое вещество (Tпл = 113.0°С; спектр ЯМР 31P – синглет с δP = 19.9 м.д.) очищали перекристаллизацией из гексана.
4-Гидроксиметилбензоат (парабен) – белое кристаллическое вещество с характерным запахом (Ткип= 275°С, Tпл = 125°С), продукт компании “Acros”, CAS 99-76-3.
4-Аллил-2-метоксифенол (эвгенол) – бесцветная, желтеющая на воздухе жидкость с сильным запахом гвоздики (Ткип = 252.7°C), очищали перегонкой под вакуумом.
Трет-бутанол – при комнатной температуре бесцветные легкоплавкие ромбические кристаллы (Tпл = 25.5°С; Ткип = 82.2°С), продукт фирмы “Компонент-реактив”.
м-Хлорнадбензойная кислота – бесцветные кристаллы (Тпл = 69‒71°C), содержание воды 3‒5%, продукт компании “Acros Organics”, CAS 937-14-4.
Натрий металлический – серебристо-белый металл (Тпл = 97°C), продукт компании “НеваРеактив”, CAS 7440-23-5.
Карбонат калия – белое кристаллическое вещество перед использованием высушивали нагреванием под вакуумом.
Органические растворители очищали согласно известным методикам.
Методики синтеза
Синтез трис-(4-метил-карбоксифенокси)-трис-(4-аллил-2-метоксифенокси)циклотрифосфазена (IIIа) с первоначальным введением парабена. В трехгорлую колбу объемом 250 мл, снабженную механическим перемешивающим устройством, термометром и обратным холодильником, загружали 5.0 г (0.0144 моля) гексахлорциклотрифосфазена, 6.57 г (0.0431 моля) парабена, 5.96 г (0.0431 моля) карбоната калия и 100 мл ацетона. Реакцию вели в течение 2 ч при 64°С и постоянном перемешивании, получая раствор соединения I (схема).
В отдельной колбе готовили фенолят эвгенола на основе 7.08 г (0.0431 моля) эвгенола и 0.99 г (0.0431 моля) металлического натрия в 50 мл диоксана. После полного растворения натрия раствор натриевой соли эвгенола добавляли к ранее подготовленному раствору I и перемешивали смесь при 102°С в течение 8 ч.
По окончании процесса реакционную смесь фильтровали, растворители удаляли при пониженном давлении и обрабатывали остаток избытком дистиллированной воды для удаления хлорида натрия и непрореагировавшего фенолята. Продукт растворяли в CH2Cl2, сушили безводным сульфатом магния, растворитель отгоняли, остаток досушивали в вакууме до постоянной массы. Выход соединения IIIа составил 10.67 г (70%).
Синтез трис-(4-аллил-2-метоксифенокси)-трис-(4-метилкарбоксифенокси)циклотрифосфазена (IIIб). В трехгорлую колбу объемом 250 мл, снабженную механическим перемешивающим устройством, термометром и обратным холодильником вводили 150 мл диоксана, 7.08 г (0.04310 моля) эвгенола и 0.99 г (0.0431моля) металлического натрия. Реакционную массу перемешивали при комнатной температуре до полного растворения натрия и добавляли 5 г (0.0144 моля) гексахлорциклотрифосфазена. Реакционную смесь нагревали до 102°С и перемешивали в течение 5 ч.
По окончании реакции полученную смесь фильтровали, растворитель удаляли при пониженном давлении и остаток сушили в вакууме до постоянной массы, выход соединения II равен 82%.
Полученное соединение II растворяли в 100 мл ацетона и добавляли натрий-парабен 12.5 г (0.0718 моля). Реакцию вели при 64°С в течение 8 ч. Образовавшуюся смесь фильтровали, растворитель удаляли при пониженном давлении и обрабатывали остаток избытком дистиллированной воды для удаления хлорида натрия и избытка фенолята. Продукт растворяли в CH2Cl2, осушали безводным сульфатом магния, растворитель отгоняли и после выдерживания в вакууме получили 8.8 г соединения IIIб с выходом 75%.
Синтез трис-(4-карбоксифенокси)-трис-(4-аллил-2-метоксифенокси)циклотрифосфазена (IV). В трехгорлую круглодонную колбу, снабженную механическим перемешивающим устройством и обратным холодильником, загружали 23.6 г трет-бутоксида натрия (0.2454 моля) и 150 мл ТГФ. Полученный раствор охлаждали до 0°C.
Второй раствор готовили в конической колбе, используя 5 г соединения IIIа (0.00464 моля) и 50 мл ТГФ.
К первому раствору добавляли 1 мл дистиллированной воды и затем вводили второй раствор, реакционную смесь перемешивали 2 ч при комнатной температуре.
По окончании реакции смесь высаждали в 1000 мл воды при перемешивании и капельном добавлении HCl до кислой среды. Осадок отфильтровывали и многократно промывали дистиллированной водой, после чего сушили в вакууме до постоянной массы.
Продукт очищали многократным промыванием дистиллированной водой. Выход соединения IV составил 58% (2.8 г).
Синтез трис-(4-эпокси-2-метоксифенокси)-трис-(4-метилкарбоксифенокси)циклотрифосфазена (V). В двугорлую колбу, снабженную дефлегматором и магнитной мешалкой, загружали 2 г (0.00186 моля) соединения IIIб и растворяли его в 10 мл хлористого метилена. К раствору при перемешивании добавляли по каплям 3.72 г (0.021 моля) м-хлорнадбензойной кислоты предварительно растворенной в 20 мл хлористого метилена. Реакцию вели при 25°С в течение 40 ч, затем реакционную массу промывали водным раствором сульфита натрия, соды, затем дистиллированной водой и сушили прокаленным сульфатом магния. После отгонки хлористого метилена продукт сушили при 40°С в вакууме до постоянной массы. Получили 1.4 г соединения V в виде желтого твердого продукта, выход 72%.
Методы исследования
Спектры ЯМР 31P и ЯМР 1H снимали при 25°C на спектрометре “Bruker CXP– 360” в различных растворителях (ацетон, диоксан, d-хлорформ) при частоте 81 и 200 МГц соответственно. В качестве внутренних стандартов использовали сигналы растворителей, химические сдвиги рассчитывали относительно стандартов – тетраметилсилана (ЯМР 1Н) и 80%-ной фосфорной кислоты (ЯМР 31Р).
Масс-спектрометрический анализ (MALDI-TOF) проводили на приборе “Bruker Auto Flex II”.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Условия реакций, выход и основные параметры спектров ЯМР 31P представлены в табл. 1. Минимальный выход (58%) наблюдается при синтезе соединения IV обработкой IIIa трет-бутанолом натрия.
Таблица 1.
Условия синтеза, выход продуктов и данные их спектров ЯМР 31P
| Соединение | Условия синтеза | Выход, % | ЯМР 31P δP, м.д. | ||
|---|---|---|---|---|---|
| растворитель | T,°C | время, ч | |||
| I | Ацетон | 64 | 2 | 86 | 17‒18 (основной синглетный сигнал) |
| II | Диоксан | 102 | 5 | 82 | То же |
| IIIа | Диоксан | 102 | 8 | 70 | 10.4 |
| IIIб | Ацетон | 64 | 8 | 75 | 10.4 |
| IV | ТГФ | 25 | 3 | 58 | 9.0 |
| V | CH2Cl2 | 25 | 40 | 72 | 9.8 |
Промежуточные триарилокситрихлорциклотрифосфазены I и II представляют собой смеси ди-, три- и тетразамещенных фосфазеновых циклов в которых, по данным спектров ЯМР 31P (рис. 1), преобладают тризамещенные соединения с соответствущим синглетным сигналом в области δP = = 17‒18 м. д. Интенсивность сигналов систем А2В и АВ2, относящихся к ди- и тетразаменным арилоксихлорциклотрифосфазенам, суммарно не превышает 10%.
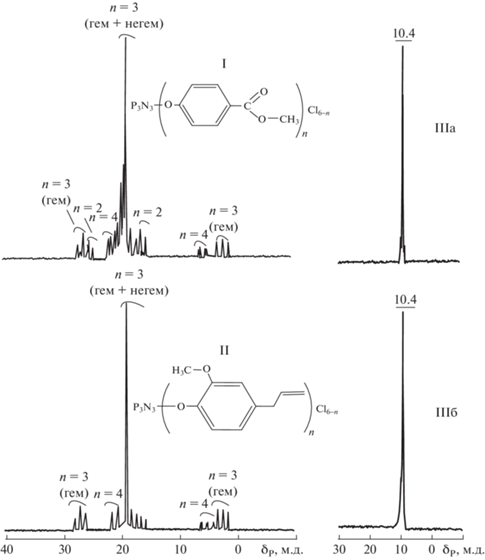
Последующее замещение остаточных атомов хлора в соединениях I и II на другие фенолы (EvgOH в случае IIIa и ParOH при синтезе IIIб) протекает в мягких условиях с образованием полностью замещенных циклических структур.
Спектр ЯМР 31P соединений IIIa и IIIб полностью идентичны и содержат синглетный сигнал при δp = 10.4 м.д., что свидетельствует об отсутствии побочных превращений. В то же время лазерные масс спектры соединения IIIа и IIIб (рис. 2) несколько различаются. В случае реакции промежуточного соединения I с EvgONa образующийся продукт IIIа содержит три соединения общей формулы P3N3(OPar)n(OEvg)6 –n с n = 4 (≈ 7%), n = 3 (51% вместе с Na+) и n = 2 (≈ 42%). MALDI-масс-спектр продукта взаимодействия II с ParONa (синтез соединения IIIб) содержит всего четыре пика, соответствующих двум соединениям с включением катионизированных ионом Na+ форм указанной выше формулы с n = 3 (43%) и n = 2 (57%).

Химические превращения функциональных групп проводили на примере соединений IIIа обработкой его трет-бутилатом Na (синтез соединения IV) или эпоксидированием алильных групп м-хлорнадбензойной кислотой (синтез соединения V) по известным методикам. Перевод сложноэфирных групп в карбоксильные во фрагментах ParO проходит с образованием преимущественно двух соединений общей формулы P3N3(PhCOOH)n(OEvg)6 –n, n = 3 (≈24%) и n = 2 (≈76%).
Необычным здесь является увеличенное количество эвгенольных радикалов в исходном соединении IIIа и образовавшемся из него продукте IV с 57 до 76% за счет понижения доли парабеновых остатков с 43 до 24 мас. %. Данный факт может быть связан с побочными превращениями сложноэфирных групп при их реакции с трет-бутилатом натрия и уменьшениям относительной доли карбоксильных групп в ароматических радикалах. Это предположение подтверждает пониженный выход соединения IV (58%, табл. 1) по сравнению с расчетным.
Нельзя также исключить возможность замены части парабеновых фрагментов на эвгенольные реакцией переарилирования, при этом значения сигналов атомов фосфора в фосфазеновых циклах не изменяются и составляют ~9.8 м.д (рис. 3).

Более сложным является состав продукта V, полученного окислением пропиленовых групп в соединении IIIб м-хлорнадбензойной кислотой (рис. 4). Как видно из табл. 2 в составе соединения V в большинстве случаев присутствует три типа соединений, большинство которых содержит 2‒3 остатка парабена, 1‒4 неконвертированных эвгенольных радикала и от 1 до 4 эпоксидных групп. Вместе с тем в соединении V содержится до 14% продуктов присоединения к эпоксидным группам воды и образовавшейся м-хлорбензойной кислоты.
Рис. 4.
MALDI-TOF масс-спектры соединений IV (а) и V (б, в). Продукт V получен при мольном соотношении IIIб : надкислота = 1 : 6 (б) и 1 : 12 (в). Значения радикалов Ar и Ar' указаны в табл. 2.

Таблица 2.
Основные соединения в продуктах V по данным MALDI-TOF масс-спектрометрии
| Значения m/z | Число остатков фенолов в формулах соединений | Относительное содержание (%) соединений в смеси при мольном соотношении IIIб : надкислота | ||||
|---|---|---|---|---|---|---|
| найдено | расчитано по формулам соединений | |||||
| ParO | EvgO | Ar* | 1 : 6 | 1 : 12 | ||
| 1078 | 1077 | 3 | 3 | ‒ | 6 | ‒ |
| 1090 | 1089 | 2 | 4 | ‒ | 8 | ‒ |
| 1094 | 1093 | 3 | 2 | 1 | 4 | ‒ |
| 1110 | 1109 | 3 | 1 | 2 | 14 | ‒ |
| 1122 | 1121 | 2 | 2 | 2 | 7 | ‒ |
| 1126 | 1125 | 3 | ‒ | 3 | 15 | 38 |
| 1148** | 1148 | 3 | 3 | ‒ | 4 | ‒ |
| 1138 | 1137 | 2 | 1 | 3 | 17 | ‒ |
| 1160** | 1160 | 2 | 1 | 3 | 5 | ‒ |
| 1154 | 1153 | 2 | ‒ | 4 | 15 | 48 |
| 1176** | 1176 | 2 | ‒ | 4 | 5 | ‒ |
| 1282‒1309 | Побочные соединения *** | ‒ | до 14% | |||



Протекание указанных побочных реакций было установлено ранее при эпоксидировании эвгенольных производных гексахлорциклотрифосфазена и высших циклофосфазенов [10].
Из данных табл. 2 можно сделать следующие заключения. Во-первых, при увеличении мольного избытка м-хлорнадбензойной кислоты: IIIб до 12 : 1 в составе обнаружено два основных соединения с m/z = 1127 и 1155, содержащие три и четыре эпоксидные группы соответственно.
Во-вторых, в большем избытке надкислоты в составе образующегося соединения V в его составе появляются до 14% указанных выше продуктов побочных превращений. Это обусловлено, с одной стороны, наличием в реакционной смеси большего количества воды, вносимой вместе с надкислотой, а, с другой, ‒ более высоким содержанием эпоксидных групп (табл. 2).
Аналогично избыток надкислоты оказывает влияние на взаимодействие эпоксигрупп с образующейся при окислении м-хлорбензойной кислотой.
Соединения IV и V с карбоксильными и эпоксидными группами могут быть использованы для модификации связующих полимерных композиционных материалов с целью регулирования физико-химических и механических характеристик последних, а также для улучшения огнестойкости. Например, самоотвержденная при 200°С равновесовая смесь IV и V с количественным содержанием гель-фракции содержит более 8% фосфора и является полностью негорючей.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации в рамках государственного задания (проект № FSSM-2022-0010).
Список литературы
Medici A., Fantin G., Pedrini P., Gleria M., Minto F. // Macromolecules. 1992. V. 25. № 10. P. 2569.
Alekperov D., Shirosaki T., Sahurai T., Popova G., Kireev V., Ihara H. // Polym. J. 2003. V. 35. № 5. P. 417.
Chandrasekhar V. Inorganic and Organometallic Polymers. Berlin: Springer Verlag, 2005.
Andrianov A.K. Polyphosphazenes for Biomedical Applications. Hoboken: Wiley, 2009.
Jaeger R. De., Gleria M. Phosphazenes: A Worldwide Insight. New York: Nova Sci. Publ., 2011. 2nd Quarter.
Kireev V.V., Chistyakov E.M., Filatov S.N., Borisov R.S., Prudskov B.M. // Polymer Science B. 2011. V. 53 № 7‒8. P. 412.
Liu J., Tang J., Wang X., Wu D. // RSC Adv. 2012. V. 2. № 13. P. 5789.
Terekhov I.V., Filatov S.N., Chistyakov E.M., Bori-sov R.S., Kireev V.V. // Russ. J. Appl. Chem. 2013. V. 86. № 10. P. 1600.
Sirotin I.S., Bilichenko Yu.V., Solodukhin A.N., Kireev V.V., Buzin M.I., Borisov R.S. // Polymer Science B. 2013. V. 55. № 5‒6. P. 241.
Сиротин И.С. Дис. … канд. хим. наук. М.: РХТУ им. Менделеева, 2013.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)