

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 6, стр. 407-416
НОВЫЕ ПОЛИХИНОЛИНЫ: СИНТЕЗ И СВОЙСТВА
М. Я. Гойхман a, *, И. В. Подешво a, Н. Л. Лорецян a, А. В. Пенькова b, А. И. Кузьминова b, И. А. Валиева a, И. В. Гофман a, А. В. Якиманский a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
b Санкт-Петербургский государственный университет
198504 Санкт-Петербург, Петродворец, Университетский пр., 26, Россия
* E-mail: goikhman@hq.macro.ru
Поступила в редакцию 15.08.2022
После доработки 15.10.2022
Принята к публикации 07.11.2022
- EDN: FHZGLW
- DOI: 10.31857/S230811392270022X
Аннотация
Синтезированы новые полихинолины, содержащие в основной цепи фрагменты 6,6'-оксидихинолина и различающиеся природой боковых сложноэфирных групп. Полученные исходные соединения и полимеры исследованы методами ИК- и ЯМР-спектроскопии, ВЭЖХ, синхронного термического (термогравиметрического и дифференциального термического) анализа, сканирующей электронной микроскопии, оценены их механические характеристики, степень набухания и углы смачивания. Из всех синтезированных полимеров сформированы диффузионные мембраны, которые были протестированы при первапорационном разделении смеси изопропиловый спирт–вода. Показано, что структура, физико-химические и транспортные свойства мембран на основе синтезированных полиамидов в значительной степени зависят от наличия в бихинолиновых фрагментах карбоксильных, а также сложноэфирных метокси- и этоксигрупп.
Известно, что наличие разветвленных структур в полимерных молекулах оказывает существенное влияние на реологические, механические и мембранные характеристики полимеров. В связи с этим среди материалов, используемых для различных приложений, все большее значение приобретают полимеры сложной архитектуры – привитые сополимеры [1], дендримеры [2], полимерные щетки [3], гребнеобразные полимеры [4].
Одним из наиболее широко используемых методов создания полимерных щеток или гребнеобразных полимеров является метод полимераналогичных превращений [5]. Однако этот метод имеет ряд серьезных недостатков. Основной из них – сложность регулирования протекания реакции химической модификации полимера, что неизбежно приводит к непостоянству содержания боковых групп. Этого недостатка можно избежать, если вместо модификации полимерной цепи проводить предварительную модификацию мономеров, из которых на следующем этапе можно формировать полимерную цепь, вводя указанные мономеры в строго заданном количестве.
Необходимо подчеркнуть, что многие отрасли современной химической промышленности требуют использования особо чистых индивидуальных соединений. В связи с этим в современной химической технологии важную роль играют процессы разделения смесей, для которых применяются традиционные неэкологичные и энергозатратные методы. Среди них следует выделить ректификацию, дистилляцию, экстракцию и адсорбцию [6–8]. В настоящее время мембранные технологии служат хорошей альтернативой данным процессам разделения в связи с их отличными эксплуатационными характеристиками: высокая эффективность разделения без добавления промежуточных токсичных реагентов, экологичность, низкая энергозатратность, простота в управлении и автоматизации процесса [9–12]. Одним из наиболее важных мембранных методов для разделения жидких смесей низкомолекулярных веществ является первапорация [13, 14].
Мембраны на основе полимеров с бихинолиновыми звеньями в основной цепи активно исследуются в первапорационных процессах [15]. Это связано с тем, что хинолиновые звенья способны к образованию комплексов с переходными металлами, например Cu (I). Такое комплексообразование позволяет регулировать морфологию мембран и, как следствие, транспортные свойства. Кроме того, морфологию мембран на основе бихинолиновых полимеров можно регулировать с помощью введения в бихинолиновые мономеры боковых групп разного строения. Это возможно, поскольку бихинолины, полученные по реакции Пфитцингера [16–18], всегда содержат карбоксильные группы, по которым можно осуществлять химическую модификацию. Нам представлялось интересным определить влияние морфологии бихинолиновых полимеров с боковыми карбоксильными и сложноэфирными группами на транспортные свойства сформированных на их основе мембран.
Цель настоящей работы – синтез новых полимеров, содержащих в основной цепи фрагменты 6,6'-оксидихинолина и различающихся длиной боковых сложноэфирных групп для исследования перспективности их применения в качестве мембранного первапорационного материала для дегидратации изопропилового спирта, а также изучение этих полимеров методами ИК- и ЯМР-спектроскопии, ВЭЖХ, синхронного термического анализа (совместно проводимых ТГА и ДТА), сканирующей электронной микроскопии (СЭМ), механических характеристик в режиме одноосного растяжения, измерение степени набухания иммерсионным методом и углов смачивания.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Очистка исходных соединений и растворителей
Хлоральгидрат, гидроксиламин гидрохлорид, сульфат натрия, концентрированная соляная кислота, концентрированная серная кислота, гидроксид калия, 4-аминоацетофенон, диэтиловый эфир, 3-метокси-4-оксибензойная кислота (ванилиновая кислота), пропиленоксид – коммерческие продукты (товарные продукты фирмы “Aldrich”), их использовали без дополнительной очистки. Другие реагенты и растворители применяли после предварительной очистки. 4,4'-диаминодифениловый эфир (ДАДФЭ) очищали перекристаллизацией из этилового спирта, Тпл = = 190°С; тионилхлорид очищали простой перегонкой, Ткип = 76°С; N-метилпирролидон (МП), Ткип = 202°С, $n_{D}^{{20}}$ = 1.4692, осушали гидридом кальция и перегоняли под вакуумом; этанол очищали простой перегонкой, Ткип = 78°С.
Синтез промежуточных соединений и мономеров
Синтез N,N'-[4,4'-бис-(4,1-фенилен)]-бис-[2-(гидроксиимино) ацетамид]оксида (1). Синтез соединения 1 проводили по методике, описанной в работе [19], для чего готовили три раствора.
Раствор 1. В круглодонную трехгорлую колбу объемом 2 л, снабженную мешалкой и термометром, помещали 950 мл воды, 275.6 г (1.941 моля) безводного сульфата натрия и 45 г (0.272 моля) хлоральгидрата. Смесь перемешивали при 40°C до полного растворения компонентов.
Раствор 2. В стакан объемом 250 мл помещали 150 мл воды, 21.3 мл концентрированной соляной кислоты и 25 г (0.125 моля) ДАДФЭ. Смесь перемешивали до полного растворения ДАДФЭ.
Раствор 3. В стакан объемом 400 мл помещали 250 мл воды и 55 г (0.797 моля) солянокислого гидроксиламина и перемешивали до полного его растворения.
При перемешивании при комнатной температуре к раствору 1 добавляли раствор 2, через ~1 мин добавляли раствор 3. Содержимое колбы нагревали до кипения и выдерживали при этой температуре 2 мин, после чего колбу охлаждали под струей воды и оставляли на ночь. Выпавший осадок отфильтровывали и промывали 750 мл воды. Продукт очищали перекристаллизацией из 1.5 л воды. Выход соединения 1 составил 39.3 г (92%).
ЯМР 1Н (ДМСО-d6), δ (м.д.): 6.99 (4Н, д, CArH); 7.67 (2Н, с, СH); 7.69 (4H, д, CArH); 10.2 (2Н, с, NH); 12.2 (2H, с, NОH).
Синтез 5,5'-оксидииндолин-2,3-диона (2) (схема 1). В круглодонную трехгорлую колбу объемом 100 мл, снабженную мешалкой и термометром, помещали 57 мл концентрированной серной кислоты, нагревали до 55°С и медленно добавляли 13.7 г (0.049 моля) тщательно высушенного и мелкоизмельченного порошка 1 так, чтобы температура реакционной смеси не поднималась выше 65°С. После введения соединения 1 температуру поднимали до 80°С, выдерживали при этой температуре 15 мин при перемешивании и оставляли охлаждаться до комнатной температуры. После охлаждения реакционную смесь выливали в керамический стакан с измельченным льдом (600 г). Выпавший красный осадок отфильтровывали, промывали водой до нейтральной реакции и сушили на воздухе при комнатной температуре. Выход соединения 2 равен 10.4 г (84%).

ЯМР 1Н (ДМСО-d6), δ (м.д.): 6.95 (2Н, д, CArH); 7.12 (2Н, с, CArH); 7.33 (2Н, д-д, CArH), 11.05 (2Н, с, NH).
Синтез 6,6'-оксибис-(2-(4-аминофенил)хинолин)-4-карбоновой кислоты) (3) (схема 1). В одногорлую круглодонную колбу, снабженную обратным холодильником, помещали раствор 6.4 г гидроксида калия в 12.5 мл воды, затем присыпали 5.3 г (0.017 моля) соединения 2, перемешивали при комнатной температуре до полного растворения, добавляли 9.4 г (0.070 моля) 4-аминоацетофенона, 39 мл этилового спирта и кипятили смесь в течение 14 ч. В охлажденную реакционную смесь выливали 50 мл воды, упаривали раствор до объема 60 мл, охлаждали, дважды экстрагировали диэтиловым эфиром, водный слой после упаривания остатков эфира подкисляли раствором соляной кислоты до рН 4. Выпавший осадок темно-красного цвета отфильтровывали и промывали водой до нейтральной реакции. Сушили. Выход 4.97 г (53%).
ЯМР 1Н, ДМСО-d6, δ, м.д.: 6.93 (4Н, д, CArH); 7.71 (2Н, д, CArH); 8.12 (4Н, д, CArH); 8.20 (2H, д, CArH); 8.33 (2Н, с, CArH); 8.43 (2H, с, CArH).
По данным элементного анализа для C32H22N4O5: вычислено, %: C – 70.84, H – 4.09, N – 10.33, O – 14.74; найдено, %: C – 70.84, H – 4.04, N – 10.38.
Синтез диметилового эфира 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновой кислоты) (4) (схема 1). В одногорлую круглодонную колбу, снабженную обратным холодильником, помещали 0.4 г (0.7 ммоля) соединения 3, 8 мл метанола и 0.4 мл концентрированной серной кислоты. Реакционную смесь кипятили 14 ч, после охлаждения до комнатной температуры выливали в 50 мл воды. Выпавший осадок отфильтровывали, промывали водой до нейтральной реакции и сушили под вакуумом до постоянной массы. Получившийся продукт перекристаллизовывали из этанола. Выход 0.33 г (78%).
ЯМР 1Н, ДМСО-d6, δ, м.д.: 3.90 (6Н, с, СH3); 6.83 (4Н, д, CArH); 7.70 (2Н, д, CArH); 8.05 (4Н, д, CArH); 8.15 (2H, д, CArH); 8.35 (2Н, с, CArH); 8.46 (2H, с, CArH).
По данным элементного анализа для C34H26N4O5: вычислено, %: C – 71.57, H – 4.59, N – 9.82, O – 14.02; найдено, %: C – 71.55, H – 4.60, N – 9.80.
По аналогичной методике получали диэтиловый эфир 5 и дибутиловый эфир 6 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновой кислоты) (схема 1).
Соединение 5. ЯМР 1Н, ДМСО-d6, δ, м.д.: 1.20 (6Н, т, СН3); 4.35 (4Н, к, СH2); 6.75 (4Н, д, CArH); 7.70 (2Н, д, CArH); 8.05 (4Н, д, CArH); 8.18 (4H, д, CArH); 8.41 (2Н, с, CArH); 8.49 (2H, с, CArH).
По данным элементного анализа для C36H30N4O5: вычислено, %: C – 72.23, H – 5.05, N – 9.36, O – 13.36; найдено, %: C – 72.28, H – 5.00, N – 9.36.
Соединение 6. ЯМР 1Н, ДМСО-d6, δ, м.д.: 0.75 (6Н, т, СН3); 1.25 (4Н, м, СH2); 1.5 (4Н, м, СH2); 4.28 (4Н, т, СH2); 6.75 (4Н, д, CArH); 7.72 (2Н, д, CArH); 8.05 (4Н, д, CArH); 8.15 (2H, с, CArH); 8.25 (2Н, д, CArH); 8.35 (2H, с, CArH).
По данным элементного анализа для C40H38N4O5: вычислено, %: C – 73.38, H – 5.85, N – 8.56, O – 12.22; найдено, %: C – 73.33, H – 5.85, N – 8.61.
Дихлорангидрид терефталоил-бис-(3-метокси-4-оксибензойной) кислоты синтезировали в соответствии с методикой, приведенной в работе [20].
Синтез полиамидов
К раствору 0.25 г (0.461 ммоля) 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновой кислоты) в 8.2 мл N-метил-2-пирролидона, охлажденному до –15oС добавляли 0.24 г (0.477 ммоля) дихлорангидрида терефталоил-бис-(3-метокси-4-оксибензойной) кислоты.
Суспензию перемешивали при –15°С в течение 60 мин, после чего снимали охлаждающую баню, добавляли 0.1 мл окиси пропилена и после полного растворения дихлорангидрида и увеличения вязкости раствора перемешивали при комнатной температуре в течение 4–5 ч.
Методика приведена для полимера I, содержащего карбоксильные группы в хинолиновых звеньях.
ИК-спектр, ν, см–1: 3500–3200 (N–H); 3200–2500 (широкая полоса колебаний связанных групп ОH); 2200–1800 (область обертонов и составных частот, характерных для ароматических структур); 1760–1735 (С=О, ароматический сложный эфир); 1690–1630 (Амид I); 1600–1510 (Амид II); 1270–1210 (С–O–C, ароматический простой эфир); 1070–1050 (С–O–C, алифатический простой эфир); 1100–1000 и 900–670 (С–H, внеплоскостные деформационные колебания хинолина).
По аналогичной методике синтезировали полимер II на основе диметилового эфира 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновой кислоты).
ИК-спектр, ν, см–1: 3500–3200 (N–H); 2830–2815 (С–H, метильная группа в сложном эфире); 2200–1800 (область обертонов и составных частот, характерных для ароматических структур); 1760–1735 (С=О, ароматический сложный эфир); 1690–1630 (Амид I); 1600–1510 (Амид II); 1270–1210 (С–O–C, ароматический простой эфир); 1070–1050 (С–O–C, алифатический простой эфир); 1100–1000 и 900–670 (С–H, внеплоскостные деформационные колебания хинолина).
Аналогично синтезировали полимер III на основе диэтилового эфира 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновой кислоты).
ИК-спектр, ν, см–1: 3500–3200 (N–H); 2900–2850 (С–H, этильная группа в сложном эфире); 2200–1800 (область обертонов и составных частот, характерных для ароматических структур); 1760–1735 (С=О, ароматический сложный эфир); 1690–1630 (Амид I); 1600–1510 (Амид II); 1270–1210 (С–O–C, ароматический простой эфир); 1070–1050 (С–O–C, алифатический простой эфир); 1100–1000 и 900–670 (С–H, внеплоскостные деформационные колебания хинолина).
Из полученных растворов полимеров в МП на стеклянных подложках отливали пленки, которые сушили при температуре 140°С до постоянной массы. Толщина пленок 30–40 мкм.
Спектры ЯМР 1Н для 1%-ных растворов регистрировали на спектрометре “Аvance-400” (“Вruker”, Германия) с рабочей частотой на ядрах 1Н – 400 МГц, внутренний стандарт Me6Si2.
Элементный анализ проводили на приборе “185B Carbon Hydrogen Nitrogen Analyzer Hewlett Packard”.
Среднюю ММ полимеров определяли методом ВЭЖХ на хроматографе фирмы “Agilent Technologies 1260 Infinity” (The Agilent 1260 Infinity Multi-Detector GPC/SEC System), снабженном тремя детекторами: рефрактометрическим (DRI, Wavelength 660 нм, простая калибровка с использованием в качестве стандарта ПММА), вискозиметрическим (VS, универсальная калибровка с использованием в качестве стандарта ПММА) и светорассеивающим (LS: Rayleigh scattering angles 150 and 900; Laser wavelength/power 660 нм/50 мВ). Температура детекторов 40°С. В установку входил набор последовательно соединенных хроматографических колонок: предколонка PLgel 5 мкм Guard 50 × 7.5 мм и две стирогелевые колонки Agilent Technologies PLgel 5 мкм MIXED-C, 300 × × 7.5 мм. Колонки термостатировали при 50°С. Подвижная фаза: 0.1 M раствор LiBr в ДМФА. Скорость подвижной фазы 1.0 мл/мин. Ввод пробы осуществляли с помощью автосамплера, объем пробы 100 мкл. Образцы полимеров (с = = 1.5 мг/мл) растворяли в элюенте.
Термический анализ образцов пленок выполняли на установке для синхронного ТГА и ДТА DTG-60 (“Shimadzu”). Образцы нагревали со скоростью 5 град/мин. Исследования проводили в воздушной атмосфере.
Механические характеристики пленок сополиамидов при комнатной температуре определяли в режиме одноосного растяжения с помощью универсальной установки для механических испытаний “AG-100kNX Plus” (“Shimadzu”, Япония). Образцы в виде полос с размерами рабочей зоны 25 × 2 мм растягивали до разрушения со скоростью 2 мм/мин в соответствии с указаниями ASTM D882-18. В процессе испытаний определяли следующие характеристики пленок: модуль упругости Е, предел пластичности σп, прочность σр и предельную деформацию до разрушения εр. Испытания каждого материала проводили на семи образцах с усреднением результатов.
ИК-спектры регистрировали при 25°С с разрешением 1 см–1 на ИК-фурье-спектрометре “BRUKER-TENSOR 27”.
Структуру поперечного скола полимерных мембран изучали методом СЭМ на микроскопе “Zeiss Merlin SEM” (“Carl Zeiss SMT”, Германия) с использованием вторичных электронов при напряжении 1 кВ. Мембраны предварительно погружались в жидкий азот для получения микрофотографии поперечного скола.
Изменение гидрофильно-гидрофобных свойств поверхности полимерных пленок в зависимости от химической структуры анализировали, измеряя углы смачивания водой методом сидячей капли с применением оптического тензиометра модели “Theta Lite” (“Biolin Scientific”). Программу OneAttension использовали для расчета характеристик капли.
Исследование равновесного набухания мембран проводили иммерсионным методом (гравиметрическим способом), в котором пленку помещали в бюкс с растворителем, выдерживали до постоянного значения массы с весовым контролем процесса. После окончания эксперимента мембраны высушивали в течение 24 ч в сушильном шкафу при 40°С и определяли их массу. В полимерных пленках было отмечено содержание остаточного растворителя (масса мембраны после сушки была меньше массы мембраны до проведения эксперимента), в связи с чем степень равновесного набухания рассчитывали по формуле
где mн – масса набухшей мембраны (г), mп – масса образца после сушки (г).Первапорационный эксперимент проводили на лабораторной установке непроточного типа с эффективной площадью мембраны 9.6 см2 при постоянном перемешивании исходной смеси при комнатной температуре (22°C) в вакуумном режиме (остаточное давление под мембраной менее 10–1 мм ртутного столба). Состав пермеата определяли при помощи газового хроматографа “Хроматэк-Кристалл” с детектором по теплопроводности и насадочной колонки, заполненной сорбентом PoraPak R1.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
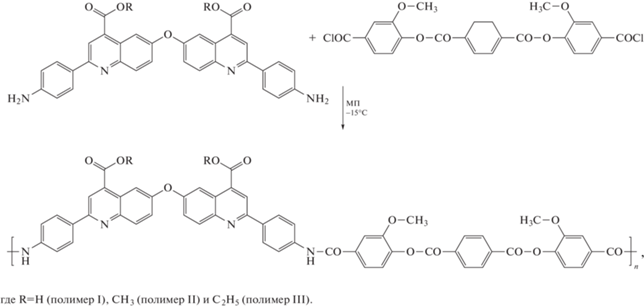
На начальном этапе работы были синтезированы реакционноспособные бифункциональные мономеры для поликонденсации, содержащие хинолиновые фрагменты – 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновая кислота) (схема 1) и ее диметиловый, диэтиловый и дибутиловый эфиры. Синтез близких по структуре соединений, а именно хинолинов, содержащих п-аминофениленовые группы, из изатина и п-аминоацетофенона описан в литературе [21, 22].
На основе указанных мономеров и дихлорангидрида терефталоил-бис-(3-метокси-4-оксибензойной) кислоты методом низкотемпературной поликонденсации синтезировали полиамиды, содержащие в основной цепи хинолиновые группы (схема 2). Дихлорангидрид терефталоил-бис-(3-метокси-4-оксибензойной) кислоты был выбран в качестве сомономера, поскольку, как было показано ранее, на его основе можно получать полиамиды с высокой характеристической вязкостью, пленки которых обладают хорошей термостойкостью и высокими механическими характеристиками [20]. Необходимо отметить, что полимер на основе дибутилового эфира 6,6'-оксибис-(2-(4-аминофенил)хинолин-4-карбоновой кислоты) 6 получить не удалось вследствие гелеобразования в процессе реакции поликонденсации.
Как следует из табл. 1, все синтезированные полимеры характеризуются достаточно высокой молекулярной массой (40–50) × 103 и индексом полидиспесности, близким к двум. Рассматривая результаты испытаний исследуемых материалов, необходимо подчеркнуть, что из синтезированных полимеров удалось изготовить прочные, оптически прозрачные пленки, обладающие высокими механическими свойствами (табл. 1) и хорошей термической стабильностью.
Таблица 1.
Механические и молекулярно-массовые характеристики
| Полимер | Е, ГПа | σп, МПа | σр, МПа | εр, % | Mw × 10–3 | Mn × 10–3 | Ð |
|---|---|---|---|---|---|---|---|
| I | 5.28 ± 0.12 | 162 ± 3 | 181 ± 3 | 21 ± 2 | 40 | 19 | 2.1 |
| II | 5.78 ± 0.08 | – | 116 ± 8 | 4.6 ± 0.7 | 44 | 22 | 2.0 |
| III | 3.83 ± 0.04 | – | 72 ± 4 | 3.9 ± 0.3 | 49 | 23 | 2.1 |
Исследование механических характеристик полиамидов показало, что в целом все синтезированные полимеры I–III обладают высокими деформационно-прочностными свойствами (табл. 1, рис. 1), вполне достаточными для того, чтобы использовать указанные полимеры в качестве мембранных материалов. Следует отметить, что при переходе от полиамидов с карбоксильными группами в хинолиновом фрагменте к их аналогам со сложноэфирными группами, равно как и с увеличением длины боковых сложноэфирных групп, деформационные характеристики пленки уменьшаются, в случае самого длинного заместителя (полимер III) падает и жесткость материала. Это, по-видимому, связано с тем, что с увеличением длины бокового заместителя в материале реализуется менее совершенная система межмолекулярных взаимодействий.
Рис. 1.
Диаграммы растяжения полимерных пленок. Цветные рисунки можно посмотреть в электронной версии.

Полученные зависимости массы образцов от температуры и тепловые эффекты, зарегистрированные в процессе их нагревания (кривые ТГА и ДТА соответственно) представлены на рис. 2.
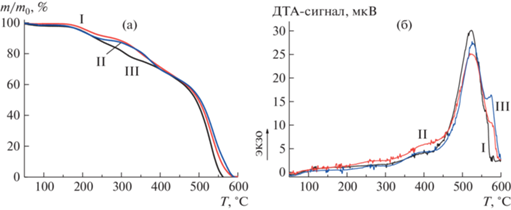
Процессы термоокислительной деструкции полимеров носят сложный, многоступенчатый характер (рис. 2). Из всех трех образцов при нагревании выше 140–150°С выделяются существенные количества низкомолекулярных примесей. Собственная деструкция полимера начинается выше 280–290°С и протекает в несколько этапов. Процессы деструкции во всех трех пленках завершаются в области температур 570–590°С полным переходом материала в газовую фазу без регистрации коксового остатка.
Температура 5% и 10%-ной потери массы (τ5 и τ10) для пленок полимеров I, II и III составляет 313, 322, 330°С и 351, 347, 358°С соответственно.
Наконец, можно констатировать, что во всем диапазоне температур, в котором проводился эксперимент (до 600°С, т.е. до полной деструкции полимера), на кривых ДТА не зарегистрированы процессы плавления. Этот факт свидетельствует о реализации для всех трех пленок неупорядоченной надмолекулярной структуры.
Внутренняя структура пленок (мембран) на основе синтезированных полимеров была изучена методом СЭМ. Микрофотографии поперечного скола мембран на основе полимеров I–III представлены на рис. 3. На них видна неоднородная структура поперечного скола всех полимерных пленок. Структура поперечного скола мембраны на основе полимера I (полиамида с карбоксильными группами в бихинолиновом фрагменте) характеризуется ярко выраженными плотно расположенными областями локализации пластической деформации, наличие которых свидетельствует о том, что даже при температуре жидкого азота разрушение материала носило частично-пластический характер. Для мембран на основе полимеров II и III (полиамиды со сложноэфирными группами) морфология меняется: для полимера II следы пластической деформации поперечного скола равномерно проявляются в форме “волнистого рельефа”. Для мембраны на основе полимера III наблюдается выраженная гетерогенность структуры поверхности скола. Таким образом, можно предположить, что переход от полиамида с карбоксильными группами в бихинолиновом фрагменте к его аналогам со сложноэфирными группами и с увеличением длины боковых сложноэфирных групп приводит к реализации более гетерогенной структуры мембран, что также подтверждается уменьшением деформационных характеристик пленок на основе полимеров II и III.

Транспорт компонентов через мембрану в первапорации описывается механизмом растворение–диффузия [23], где основными стадиями являются селективная сорбция компонентов на мембране и их диффузия через мембрану. Для оценки транспорта компонентов через мембраны и изменения гидрофильно-гидрофобных свойств поверхности мембран вследствие увеличения длины сложноэфирных групп были исследованы такие параметры, как углы смачивания водой и степень набухания в воде и спиртах (этаноле, изопропиловом спирте, метаноле) (табл. 2).
Таблица 2.
Степень набухания мембран на основе полимеров I–III в различных растворителях
| Растворитель | Степень набухания, % | ||
|---|---|---|---|
| полимер I | полимер II | полимер III | |
| Вода | 15 | 65 | 23 |
| Этанол | 14 | 75 | 24 |
| Изопропиловый спирт | 5 | 8 | 1 |
| Метанол | 17 | 88 | 16 |
Было показано, что переход от полиамида с карбоксильными группами в бихинолиновом фрагменте (полимер I) к его аналогам со сложноэфирными группами не приводит к изменению гидрофильности поверхности мембран: углы смачивания водой для всех мембран на основе полимеров I–III близки по значениям (80°–85°).
Представленные в табл. 2 данные демонстрируют высокую степень набухания мембран в воде, метаноле и этаноле. Набухание в изопропиловом спирте незначительно для всех образцов. Мембраны из полиамидов со сложноэфирными группами обладали большей степенью набухания в растворителях по сравнению с мембраной из полимера I, что связано с изменением внутренней морфологии мембран. Мембрана на основе полимера II характеризовалась наибольшим набуханием в растворителях по сравнению с мембранами из полимеров I и III (рис. 3). На основании полученных данных по набуханию смесь вода–изопропиловый спирт была выбрана для проведения первапорационного эксперимента с использованием разработанных мембран из-за наибольшей разницы степени набухания в воде и изопропиловом спирте, что обеспечит их селективное разделение (табл. 2).
Транспортные свойства полимерных мембран были изучены в процессе первапорации для дегидратации изопропилового спирта (20 мас. % воды). Разделение смеси вода–изопропиловый спирт затруднено традиционными методами, например таким, как дистилляция, в связи с тем, что изопропиловый спирт образует азеотроп с водой (вода : изопропиловый спирт = 12 : 88 мас. %, температура кипения 80.3°С [24]). Однако данное разделение может быть эффективно проведено, к тому же без добавления дополнительных реагентов, в процессе первапорации, что обусловлено спецификой массопереноса низкомолекулярных веществ по механизму растворение–диффузия, в котором сначала происходит селективная сорбция низкомолекулярного разделяемого компонента, затем его диффузия и десорбция с обратной стороны мембраны. Важную роль в данном разделении играет материал мембраны. Результаты первапорации смеси изопропиловый спирт (80 мас. %)–вода (20 мас. %) при комнатной температуре при помощи разработанных мембран представлены на рис. 4.
Рис. 4.
Транспортные свойства мембран на основе полимеров I–III при первапорации смеси изопропиловый спирт (80 мас. %)–вода (20 мас. %) при 22°С.

Приведенные данные демонстрируют селективность всех мембран по отношению к воде (более 88 мас. % в пермеате). Мембрана на основе полимера I имеет наименьшую удельную производительность (10 г/(м2 ч)) и наибольшую селективность (99.9 мас. % воды в пермеате). Это может быть обусловлено более жесткой внутренней структурой и системой межмолекулярных взаимодействий мембраны, что способствует ее меньшему набуханию в компонентах разделяемой смеси и приводит к наименьшей удельной производительности. Мембрана на основе полимера III характеризуется увеличением в 6.9 раза удельной производительностью (69 г/(м2 ч)), но с понижением содержания воды в пермеате до 88.3 мас. % по сравнению с мембраной из полимера I. Данный эффект может быть связан с изменением внутренней структуры на более гетерогенную при увеличении длины боковых сложноэфирных групп. Оптимальные транспортные свойства продемонстрировала мембрана из полимера II в процессе первапорации смеси изопропиловый спирт (80 мас. %)–вода (20 мас. %) при 22°С: производительность 95.1 г/(м2 ч), 95.1 мас. % воды в пермеате. Наивысшая удельная производительность обусловлена менее плотной структурой мембраны с наивысшими значениями степени набухания в компонентах смеси и менее совершенной системой межмолекулярных взаимодействий в набухшем состоянии (табл. 2), что также способствует незначительному снижению селективных свойств. При набухании мембраны свободный объем между цепями полимера становится больше, что вызывает совместный массоперенос через мембрану воды и изопропилового спирта, приводя к понижению содержания воды в пермеате.
Для изучения стабильности мембраны на основе полимера II, обладающей оптимальными транспортными характеристиками, были изучены ее свойства в процессе первапорации смеси изопропиловый спирт–вода в широком концентрационном диапазоне в течение двух циклов разделения исходной смеси, содержащей от 20 до 100 мас. % воды (рис. 5), с последующим возвращением к концентрации 20 мас. % воды в исходной смеси (третий цикл).
Рис. 5.
Зависимость удельной производительности (а) и содержания воды в пермеате (б) от ее содержания в исходной смеси для мембраны изопропиловый спирт–вода при первапорации через мембрану полимера II. 1, 2 и 3 – первый, второй и третий цикл первапорации соответственно.

Была отмечена стабильность транспортных характеристик при проведении нескольких циклов первапорационного эксперимента, а именно сохранение значений для удельной производительности и содержания воды в пермеате, обусловленное стабильностью мембраны и демонстрирующее перспективность ее использования в течение продолжительного времени.
Таким образом, было показано, что структура, физико-химические и транспортные свойства пленок (мембран) на основе синтезированных полиамидов, содержащих в основной цепи хинолиновые группы, в значительной степени зависят от групп в хинолиновом фрагменте (карбоксильная или сложноэфирные группы) и от увеличения длины боковых сложноэфирных групп. Полученные транспортные характеристики мембран на основе полимеров II и III со сложноэфирными группами при первапорационной дегидратации изопропилового спирта демонстрируют перспективность их применения в качестве мембранного материала.
Список литературы
Баттерд Г., Трегер Д. Свойства привитых и блок-сополимеров. Л.: Химия, 1970.
Tanaka N., Tanigawa T., Hosoya K., Kimata K., Araki T., Terade S. // Chem. Lett. 1992. V. 6. P. 959.
Milner S.T. // Science. 1991. V. 251. № 4996. P. 905.
Saha D., Peddireddy K.R., Allgaier J., Zhang W., Maccarone S., Frielinghaus H., Richter D. // Nanomaterials. 2020. V. 10. № 12. P. 2410.
Платэ Н.А., Шибаев В.П. Гребнеобразные полимеры и жидкие кристаллы. М.: Химия, 1980.
Kuzminova A.I., Dmitrenko M.E., Mazur A., Ermakov S.S., Penkova A.V. // Sustainability. 2021. V. 13. P. 6092.
Slaughter R.J., Mason R.W., Beasley D.M.G., Vale J.A., Schep L.J. // Clin. Toxicol. 2014. V. 52. P. 470.
Swapna V.P., Thomas S.P., Jose T., Moni G., George S.C., Thomas S., Stephen R. // J. Mater. Sci. 2019. V. 54. P. 8319.
Dmitrenko M.E., Penkova A.V., Missyul A.B., Kuzminova A.I., Markelov D.A., Ermakov S.S., Roizard D. // Separation Purification Technol. 2017. V. 187. P. 285.
Gao R., Zhang Q., Lv R., Soyekwo F., Zhu A., Liu Q. // Chem. Eng. Res. Des. 2017. V. 117. P. 688.
Dong Y.Q., Zhang L., Shen J.N., Song M.Y., Chen H.L. // Desalination. 2006. V. 193. P. 202.
Yushkin A.A., Golubev G.S., Podtynnikov I.A., Borisov I.L., Volkov V.V., Volkov A.V. // Pet. Chem. 2020. V. 60. P. 1317.
Dmitrenko M.E., Penkova A.V., Missyul A.B., Kuzminova A.I., Markelov D.A., Ermakov S.S., Roizard D. // Separation Purification Technol. 2017. V. 187. P. 285.
Wu J.-K., Yin M.-J., Han W., Wang N., An Q.-F. // J. Mater. Sci. 2020. V. 55. P. 12607.
Polotskaya G., Pulyalina A., Goikhman M., Podeshvo I., Gofman I., Shugurov S., Rostovtseva V., Faykov I., Tataurov M., Toikka A., Polotsky A. // Polymers. 2019. V. 11. № 10. P. 1542.
Pfitzinger W. // J. Prakt. Chem. 1897. V. 56. № 2. P. 283.
Buu-Hoi N.P., Royer R., Xuong N.D., Jacquignon P. // J. Org. Chem. 1953. V. 18. № 9. P. 1209.
Shvekhgeimer M.-G.A. // Chem. Heterocycl. Compd. 2004. V. 40. № 3. P. 257.
Valieva I.A., Goikhman M.Ya., Podeshvo I.V., Loretsyan N.L., Gofman I.V., Smyslov R.Yu., Litvinova L.S., Yakimansky A.V. // Polymer Science B. 2019. V. 61. № 3. P. 302.
Goikhman M.Ya., Yevlampieva N.P., Kamanina N.V., Podeshvo I.V., Gofman I.V., Mil’tsov S.A., Khurchak A.P., Yakimanskii A.V. // Polymer Science A. 2011. V. 53. № 6. P. 457.
Rangnekar D.W., Shenoy G.R. // Dyes Pigments. 1987. V. 8. № 4. P. 281.
Kondrashova N.N., Shvekhgeimer M.-G.A. // Dokl. Chem. 2005. V. 400. № 2. P. 21.
Мулдер М. Введение в мембранную технологию. М.: Мир, 1999.
Огородников С.К., Лестева Т., Коган В. Азеотропные смеси: Справочник. Л.: Химия, 1971.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)