

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 6, стр. 417-427
Контролируемая сополимеризация акрилонитрила и этил-2-цианоакрилата в условиях обратимой передачи цепи и термическое поведение полученных сополимеров
Р. В. Томс a, А. Ю. Гервальд a, М. С. Балашов a, Н. И. Прокопов a, А. В. Плуталова b, Е. В. Черникова b, *
a МИРЭА – Российский технологический университет, Институт тонких химических технологий
им. М.В. Ломоносова
119571 Москва, пр. Вернадского, 86, Россия
b Московский государственный университет имени М.В. Ломоносова, Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 03.11.2022
После доработки 15.11.2022
Принята к публикации 17.11.2022
- EDN: SWEATD
- DOI: 10.31857/S2308113922700255
Аннотация
Впервые синтезированы сополимеры акрилонитрила и этил-2-цианоакрилата. Показано, что в условиях радикального инициирования в присутствии 2-(додецилтиокарбонотиоилтио)-2-метилпропионовой кислоты в качестве агента обратимой передачи цепи образуются сополимеры контролируемой молекулярной массы с узким молекулярно-массовым распределением. Определена относительная активность мономеров в сополимеризации, составившая r1 = 0.052 ± 0.02 для акрилонитрила и r2 = 54.5 ± 7.2 для этил-2-цианоакрилата. Используя разные способы введения этил-2-цианоакрилата в сополимеризацию (единовременная и непрерывная загрузка), синтезированы сополимеры разной микроструктуры. Показано, что при нагревании сополимеров в инертной атмосфере протекают два конкурентных процесса – деполимеризация и циклизация нитрильных звеньев. Вероятность деполимеризации возрастает при увеличении доли этил-2-цианоакрилата в сополимере и при переходе от градиентной к статистической структуре сополимера.
ВВЕДЕНИЕ
Сополимеры акрилонитрила традиционно используют для получения текстильных и углеродных волокон [1–3]. Такие сополимеры обычно содержат разнообразные виниловые мономеры, включающие (мет)акрилаты, винилацетат, виниловые кислоты и т.д. [4–17]. Тем не менее, до сих пор продолжаются поиски новых сомономеров, которые могли бы решить сразу несколько задач: расширить температурный интервал термоокислительной стабилизации и сместить его в область более низких температур. На практике для этой цели часто используют два сомономера – ускоритель циклизации, к которым относятся акриловая, метакриловая и итаконовая кислоты, и внутренний пластификатор, например метилакрилат [18, 19]. Совместить свойства двух сомономеров в одном пока не удается.
В настоящей работе мы предположили, что этил-2-цианоакрилат (ЭЦА), содержащий одновременно нитрильную и сложноэфирную группу в одном мономерном звене, позволит решить эту проблему. Алкил-2-цианоакрилаты широко используются как цианоакрилатные клеи [20]. Сочетание двух акцепторных заместителей при одном атоме углерода обеспечивает легкость полимеризации цианоакрилатов по анионному механизму в присутствии даже слабых нуклеофилов (вода, спирты и другие) [21–26]. Описана также цвиттер-ионная полимеризация цианоакрилатов под действием ДМСО и других реагентов [21, 27, 28].
Известна радикальная полимеризация алкилцианоакрилатов под действием традиционных инициаторов – перекиси бензоила и ДАК [29–35]. Для ее проведения необходимо подавить полимеризацию по анионному механизму, что возможно в присутствии ряда сильных кислот (серной кислоты, дихлоруксусной и трифторуксусной кислоты, метансульфокислоты, 1,3-пропансультоны и т.д.). Кинетические закономерности радикальной полимеризации алкилцианоакрилатов аналогичны другим виниловым мономерам, например ММА. Однако алкилцианоакрилаты более активны в полимеризации, чем метакрилаты [36]. Так, для ЭЦА константа скорости роста при 30°С составляет ~1.6 × 103 л/моль с [32], что соответствует константе скорости роста ММА при 80°С [37].
При сополимеризации с мономерами, содержащими электронодонорные заместители, алкилцианоакрилаты образуют чередующиеся сополимеры [36, 38, 39]. Такая тенденция оказалась характерной для виниловых эфиров, винилацетата, стирола, α-метилстирола, а также для ММА. В научной и патентной литературе практически не описаны сополимеры АН с алкил-2-цианоакрилатами, за исключением дициклогексил-аммонийной соли цианоакриловой кислоты [40, 41]. В данном случае оказалось, что такой сомономер способствует расширению температурного интервала циклизации за счет наличия в структуре мономера карбоксильной группы.
Алкилцианоакрилаты полимеризуются по механизму радикальной полимеризации с обратимой деактивацией цепи [42], что было показано на примере синтеза блок-сополимера с ММА под действием цианизопропилдитиобензоата в качестве агента обратимой передачи цепи (ОПЦ). Однако авторы обнаружили, что дитиобензоатный фрагмент может отщепляться от макромолекулы в виде дитиобензойной кислоты, что приводит к нарушению ОПЦ-механизма и появлению на конце цепи связи С=С. На примере этил-, бутил- и фенилэтил-2-цианоакрилата было установлено, что доля “живых” цепей в конце полимеризации составляет ~30%.
Вместе с тем полиалкилцианоакрилаты неустойчивы при нагревании, действии ряда основных реагентов и легко деструктируют [43–47]. При этом возможна деполимеризация, которая наблюдается при температурах, немного превышающих их температуру стеклования [48]. При нагревании поли(этил-2-цианоакрилата) выше 165°С начинается деполимеризация, энергия активации данного процесса составляет 37.4 ккал/моль [49]. С ростом длины алкильного заместителя энергия активации деполимеризации возрастает [43, 50].
При сополимеризации АН с алкилцианоакрилатами можно ожидать, что в образующемся сополимере при нагревании будет происходить частичная деструкция за счет наличия звеньев алкилцианоакрилата, которая будет сопровождаться появлением радикалов, способных инициировать циклизацию нитрильных групп АН.
Таким образом, цель настоящей работы ‒ синтез новых сополимеров АН и ЭЦА в условиях ОПЦ-процесса и изучение их термического поведения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мономеры – АН (99%) фирмы “Acros” очищали перегонкой по стандартной методике, ЭЦА (98%) фирмы “Weisse” использовали без дополнительной очистки. Инициатор ДАК перекристаллизовывали из этанола, хранили в темноте при −3°С. ДМСО (99%) и ДМФА (для ВЭЖХ) фирмы “Fluka” перед применением перегоняли. ОПЦ-агент 2-(додецилтиокарбонотиоилтио)-2-метилпропионовую кислоту (ДТМПК, 99%) и ингибитор анионной полимеризации п-толуолсульфокислоту (ТСК, 99%) фирмы “Aldrich” использовали без дополнительной очистки.
Сополимеризацию АН и ЭЦА (1–40 мол. %) для оценки относительной активности мономеров проводили в ДМСО при 80°С в присутствии 0.2 мас. % ТСК в расчете на общий объем и ДАК 3 × 10–3 моль/л. Полимер выделяли на начальной конверсии (до 10%), высаживая полимеризат в смесь ацетона и гексана (40 : 60 мас. %). Полученный полимер фильтровали, промывали, сушили при температуре 60°С и анализировали состав сополимера.
Аналогичным образом проводили сополимеризацию АН и ЭЦА по механизму ОПЦ. Рассчитанное количество ДАК и ДТМПК (3 × 10–3 моль/л), а также ТСК (0.2 мас. %) растворяли в ДМСО, чтобы суммарная концентрация мономеров в растворе составляла 40 мас. %. Использовали два способа введения мономеров в синтез. В первом одновременно добавляли АН и ЭЦА, во втором сразу вводили АН, а ЭЦА добавляли с выбранной скоростью непосредственно в ходе полимеризации шприцевым насосом. Колбу с реакционной смесью продували аргоном (99.99%) в течение 15 мин, устанавливали обратный холодильник и погружали в баню, разогретую до 80°С. Температуру поддерживали с точностью измерения ±0.1°С. В ходе синтеза через заданное время отбирали пробы для оценки конверсии и анализа продукта. Реакционную смесь при необходимости разбавляли ДМСО, содержащим ТСК для предотвращения анионной полимеризации ЭЦА, высаживали в избыток воды, содержащей ТСК, сополимеры фильтровали, промывали водой и сушили при 80°С до постоянной массы. Конверсию определяли гравиметрически.
Молекулярно-массовые характеристики ПАН изучали методом ГПХ на хроматографе GPC-120 фирмы “PolymerLabs”. Анализ проводили при 50°С в ДМФА, содержащем 0.1 мас. % LiBr, со скоростью потока 1 мл/мин. Для разделения использовали две колонки PLgel 5 µm MIXED С (М = (5 × 102)– (1 × 107)). Средние ММ и ММР рассчитывали по стандартам ПММА и пересчитывали для ПАН, используя коэффициенты уравнения Марка−Куна−Хаувинка (KПАН = 39.4 × 10–4, α = 0.75, KПMMA = 17.7 × 10–4, α = 0.62 [51]).
Сополимеры анализировали в виде пленок. Для этого готовили 8%-ный раствор полимеров в ДМСО, наливали его на стеклянную горизонтальную подложку и испаряли растворитель при 80°С в течение 3 ч. Пленки удаляли и экстрагировали остаточный ДМСО водой в течение 24 ч, затем их сушили под вакуумом при комнатной температуре до постоянной массы. Готовые пленки нарезали на квадратные образцы размером 20 × 20 мм; толщина пленки составляла от 50 до 100 мкм.
Тепловые эффекты, наблюдаемые при динамическом нагревании полимеров, исследовали на дифференциальном сканирующем калориметре “Netzsch DSC 204” фирмы “Netzsch” (Германия) в атмосфере осушенного аргона при скорости потока 50 мл/мин в температурном интервале 30‒500°С со скоростью нагревания 10 град/мин. Для проведения измерений брали образцы массой от 4 до 8 мг и помещали в стандартный алюминиевый тигель. Результаты обрабатывали с помощью программы Netzsch Proteus.
Термогравиметрический анализ выполняли на термическом анализаторе “STA 449 F3 Jupiter” (“Netzsch”). Потерю массы 15–20 мг образцов сополимеров АН, помещенных в корундовый тигель, анализировали при линейном нагревании со скоростью 10 град/мин от 25 до 600°С в атмосфере аргона со скоростью потока 50 мл/мин.
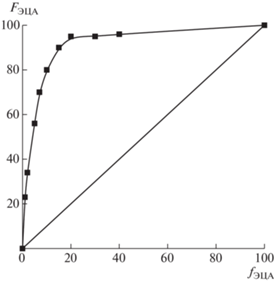
Состав сополимеров АН методом ИК-спектроскопии изучали на ИК-фурье спектрометре “Spectrum Two FT–IR Spectrometer” фирмы “Perkin Elmer” в области 4000–400 см–1. Для количественного определения состава сополимеров использовали калибровочную зависимость отношения интенсивности оптических полос A1730/A2243 от мольного отношения звеньев ЭЦА и АН в смеси гомополимеров, где A1730 и A2243 отвечают интенсивности характеристических полос поглощения валентных колебаний связей C=O (νC=O = = 1730 см–1) для ЭЦА и групп CN (νCN = 2243 см–1) для АН (рис. 1).
Рис. 1.
Калибровочная зависимость отношения характеристических полос поглощения карбонильной (A1730) и нитрильной (A2240) групп от мольного содержания звеньев ЭЦА (FЭЦА) и АН (FАН) в смеси гомополимеров для определения состава сополимера АН и ЭЦА. Цветные рисунки можно посмотреть в электронной версии.

Изменения, происходящие в структуре макромолекул при термоокислительной стабилизации в изотермических условиях, изучали методом ИК-НПВО-спектроскопии. Для этого пленку образца помещали в ячейку ДСК “Netzsch DSC 204” в инертной атмосфере и выдерживали при фиксированной температуре в течение заданного времени, затем регистрировали ИК-спектры при комнатной температуре в режиме НПВО (кристалл алмаз) в области от 4000 до 600 см–1 на ИК-спектрометре “Spectrum Two FT–IR Spectrometer” с разрешением 0.5 см–1.
Долю непрореагировавших нитрильных групп φCN [52] определяли по уравнению
(1)
${{{{\varphi }}}_{{{\text{CN}}}}} = \frac{{{{A}_{{{\text{C}} \equiv {\text{N}}}}}}}{{{{A}_{{{\text{C}} \equiv {\text{N}}}}} + f{{A}_{{ - {\text{C}} = {\text{N}} - }}}}},$РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез сополимеров акрилонитрила и этил-2-цианоакрилата и их характеристики
ЭЦА способен полимеризоваться как по радикальному, так и по анионному механизму. Последний легко реализуется в присутствии ДМСО, и для подавления анионной полимеризации ЭЦА используют кислоты [31]. Экспериментальным путем было установлено, что 0.2 мас. % ТСК в расчете на систему, содержащую 60 мас. % ДМСО, 20 мас. % АН и 20 мас. % ЭЦА, предотвращает образование полимера в течение 24 ч. Этого достаточно для приготовления реакционных смесей и проведения полимеризации по радикальному механизму.
Оценка констант сополимеризации АН и ЭЦА по схеме Q–e [51], дает значения rАН = 0.07 и rЭЦА = 13.7, что позволяет ожидать более высокую активность ЭЦА в сополимеризации с АН. Относительная активность АН и ЭЦА в радикальной сополимеризации была определена из анализа состава сополимеров, синтезированных на начальных конверсиях из мономерных смесей разного состава (рис. 2), методами наименьших квадратов [53] и Файнемана–Росса [54]. Оказалось, что ЭЦА на три порядка более активен в сополимеризации с АН: rАН = 0.052 ± 0.02 и rЭЦА = = 54.5 ± 7.2 (метод наименьших квадратов) и rАН = 0.042 ± 0.01 и rЭЦА = 46.7 ± 11.9 (метод Файнемана–Росса). Следовательно, независимо от состава мономерной смеси ЭЦА расходуется быстрее, чем АН, и на глубоких конверсиях должен образовываться сополимер с высокой композиционной неоднородностью по составу.
Рис. 2.
Диаграмма состава сополимера АН и ЭЦА, образующегося в условиях классической радикальной сополимеризации. FЭЦА и fЭЦА – мольная доля ЭЦА в сополимере и мономерной смеси в процентах.
Для устранения композиционной неоднородности сополимера можно использовать радикальную полимеризацию с обратимой деактивацией цепи, например ОПЦ-полимеризацию [55]. Вследствие огромной разницы в величинах активности мономеров в данном случае будут образовываться макромолекулы градиентного состава с высокой однородностью по молекулярной массе и составу. Для синтеза сополимеров с равномерным распределением звеньев ЭЦА вдоль цепи необходимо регулировать концентрацию ЭЦА в реакционной смеси в ходе сополимеризации. В связи с этим в настоящей работе сополимеризацию АН и ЭЦА в условиях ОПЦ-механизма проводили в двух режимах: 1) АН и ЭЦА вводили в реакционную смесь перед началом синтеза; 2) ЭЦА добавляли непрерывно в ходе полимеризации (табл. 1).
Таблица 1.
Рецептуры и условия синтезов сополимеров АН и ЭЦА в ДМСО в условиях ОПЦ-полимеризации и их характеристики
| Образец | f*ЭЦА, мол. % | VЭЦА + ДМСО, мл/ч | f**ЭЦА, мол. % | Конверсия, % | Mn × 10–3 | Đ | FЭЦА, мол. % |
|---|---|---|---|---|---|---|---|
| ПАН | 0 | Не вводили | 0 | 55 | 28.8 | 1.22 | 0 |
| 1 | 5 | Вводили сразу | 5 | 66 | 29.0 | 1.24 | 12.3 |
| 2 | 10 | Вводили сразу | 10 | 66 | 32.0 | 1.23 | 17.1 |
| 3 | 0 | 1.12 | 11.36 | 55 | 23.4 | 1.43 | 38.2 |
| 4 | 0 | 0.65 | 7.18 | 58 | 24.0 | 1.45 | 17.0 |
| 5 | 0 | 0.36 | 4.26 | 56 | 23.8 | 1.33 | 11.8 |
| 6 | 0 | 0.16 | 1.84 | 58 | 25.8 | 1.40 | 4.7 |
Независимо от способа введения ЭЦА сополимеризация протекает без индукционного периода (рис. 3), а начальная скорость сополимеризации возрастает с увеличением доли ЭЦА в системе. Значения максимально достижимой конверсии за 4.5 ч реакции при 80°С составляют 55–65% и повышаются при переходе от непрерывной к единовременной загрузке ЭЦА.
Рис. 3.
Зависимость конверсии от времени сополимеризации АН и ЭЦА в ДМСО в присутствии 3 × × 10–3 моль/л ДАК, 3 × 10–3 моль/л ДТМПК и 0.2 мас. % ТСК при 80°С. Способы введения ЭЦА: 1, 2 – единовременный, 3–6 –непрерывный; fЭЦА в исходной смеси 5 (1) и 10 мол. % (2); скорость введения ЭЦА 1.12 (3), 0.65 (4), 0.36 (5) и 0.16 мл/ч (6).

Основным признаком реализации ОПЦ-механизма является линейный рост среднечисленной молекулярной массы Mn и образование полимера с узким ММР. Во всех изученных системах наблюдаются унимодальное ММР сополимеров (рис. 4а), линейный рост Mn от конверсии (рис. 4б) и низкое значение дисперсности сополимеров Ð = 1.24–1.45 (рис. 4в). Предварительные эксперименты показали, что Mn сополимеров в исследуемых системах изменяется обратно пропорционально концентрации ОПЦ-агента. Эти результаты подтверждают реализацию ОПЦ-механизма полимеризации и доказывают, что анионный механизм полимеризации ЭЦА подавлен.
Рис. 4.
Нормированные к единичной площади кривые ММР сополимеров, образующихся при единовременной загрузке мономеров и fЭЦА = 5 мол. % в исходной смеси на разных конверсиях (а), а также зависимости Mn (б) и дисперсности Đ (в) сополимеров АН и ЭЦА от конверсии. Здесь и далее номера точек соответствуют номерам образцов в табл. 1.

Очевидно, что способ введения ЭЦА в сополимеризацию должен отразиться на составе сополимеров, образующихся на разных конверсиях. На рис. 5 приведены зависимости мольной доли ЭЦА в сополимере от суммарной конверсии мономеров для всех экспериментов. При единовременном введении АН и ЭЦА в сополимеризацию наблюдается быстрый расход ЭЦА (кривые 1 и 2), что связано с высокой разницей в активности мономеров. Изменение среднего состава сополимера в процессе сополимеризации наряду с реализацией ОПЦ-механизма указывает на формирование сополимера градиентной структуры [56]:
Рис 5.
Зависимость мольной доли ЭЦА в сополимере FЭЦА от конверсии мономеров для сополимеров АН и ЭЦА, полученных ОПЦ-сополимеризацией в ДМСО в присутствии [ДАК]0 = 3 × 10–3 моль/л, [ДТМПК]0 = = 3 × 10–3 моль/л и ТСК = 0.2 мас. % при 80°С.


При непрерывном введении ЭЦА состав сополимеров сохраняется постоянным на протяжении сополимеризации (кривые 3–6). В этом случае образуется не градиентный, а статистический композиционно однородный сополимер:

Чем выше скорость введения ЭЦА, тем больше его доля в сополимере. Состав сополимеров, выделенных на предельных конверсиях, приведен в табл. 1. Видно, что, изменяя способ и скорость введения ЭЦА, можно получить сополимеры с одинаковым средним составом, но с разной микроструктурой цепи.
Ранее мы показали, что термическое поведение сополимеров АН, свойства их растворов и механические характеристики сформованных волокон зависят от микроструктуры цепи сополимеров [57]. Следует ожидать, что в изучаемых системах тоже обнаружится такая зависимость.
Исследование термического поведения сополимеров в инертной атмосфере
Термическое поведение ПАН и поли(этил-2-цианоакрилата) в инертной атмосфере существенное различается [18, 49]. Поли(этил-2-цианоакрилат) начинает деструктировать с выделением мономера при 160°С, достигая максимума скорости деструкции при ~265°С [43, 49]. При нагревании ПАН происходит экзотермическая реакция внутри- и межмолекулярной циклизации [19], протекающая в узком интервале температур с максимумом экзо-эффекта около 276°С (рис. 6а):


Предсказать априори влияние введения звеньев ЭЦА в ПАН на протекание реакции циклизации сложно. С одной стороны, каждое мономерное звено сополимера содержит нитрильную группу и может участвовать в циклизации. С другой стороны, может доминировать деструкция полимера.
Как видно на рис. 6а и в табл. 2, экзо-эффект в сополимерах наблюдается при более высоких температурах, чем у ПАН, а интенсивность тепловыделения для них ниже. В целом с ростом содержания ЭЦА в сополимере температура максимума экзо-эффекта повышается, а тепловой эффект уменьшается. В принципе близкое поведение проявляет и полиметакрилонитрил, который подвергается деполимеризации при температуре выше 220°С, а циклизации – при наличии примесей или малого количества звеньев метакриловой кислоты [58]. Таким образом, появление в цепи АН звеньев мономеров, содержащих при четвертичном атоме углерода заместитель, отличный от водорода, приводит к смещению процессов циклизации в область более высоких температур по сравнению с ПАН.
Таблица 2.
Анализ термограмм сополимеров АН и ЭЦА, зарегистрированных в инертной атмосфере
| Образец | FЭЦА, мол. % | Мзв, г/моль | Тэндо, °С | ΔHэндо | Тэкзо, °С | –ΔHэкзо | ||
|---|---|---|---|---|---|---|---|---|
| Дж/г | Дж/моль | Дж/г | Дж/моль | |||||
| 0 | 0 | 53 | – | – | – | 276 | 828 | 15.6 |
| 1 | 12.3 | 62 | 232 | 40 | 0.6 | 311 | 653 | 10.5 |
| 2 | 17.1 | 65 | 232 | 131 | 1.9 | 315 | 573 | 8.8 |
| 3 | 38.2 | 80 | 224 | 345 | 4.3 | 322 | 378 | 4.7 |
| 4 | 17.0 | 65 | 183 | 342 | 5.0 | 308 | 469 | 7.2 |
| 5 | 11.8 | 62 | 190 | 138 | 2.2 | 314 | 597 | 9.6 |
| 6 | 4.7 | 56 | – | – | – | 295 | 759 | 13.6 |
При этом в сополимерах, полученных при единовременной загрузке мономеров, теплота циклизации в расчете на моль звена выше, чем у сополимеров того же состава, но синтезированных при непрерывном введении ЭЦА. Независимо от микроструктуры цепи теплота циклизации в расчете на моль звена оказывается ниже, чем у ПАН. Кроме того, в диапазоне 150–250°С на термограммах сополимеров регистрируется дополнительный эндотермический пик, который отсутствует у ПАН (рис. 6б, табл. 2). Тепловой эффект данного процесса в расчете на моль звена возрастает с увеличением доли ЭЦА в сополимере и при переходе от единовременного к непрерывному введению ЭЦА. Можно предположить, что данный тепловой эффект связан с деполимеризацией за счет наличия в цепи сополимера звеньев ЭЦА.
Анализ полимеров методом ТГА показал, что термостойкость синтезированных сополимеров зависит от содержания ЭЦА и распределения звеньев в цепи (рис. 7). Температура начала понижения массы у ПАН соответствует максимуму экзо-эффекта на термограмме ДСК. Статистический сополимер с 4.7 мол. % ЭЦА (кривая 6) обладает близкой к ПАН термостойкостью, хотя небольшое понижение массы наблюдается уже при температуре выше 160°С. При дальнейшем повышении доли ЭЦА в статистических сополимерах температура начала термодеструкции практически не изменяется, а на термограммах становится заметной низкотемпературная ступень потери массы, интенсивность которой увеличивается с ростом доли ЭЦА в сополимере (кривые 4 и 5). При этом появляется характерный запах ЭЦА. Заметим, что в указанной области температур наблюдается эндотермический пик на кривых ДСК. Для градиентных сополимеров низкотемпературная ступень потери массы регистрируется при температуре выше 200°С. Однако в данном случае понижение доли ЭЦА в сополимере приводит к повышению термостойкости сополимера (кривые 1 и 2).
Рис. 7.
Кривые потери массы ПАН и сополимеров АН и ЭЦА в аргоне при скорости нагревания 10 град/мин.

Обращает на себя внимание тот факт, что сополимеры градиентного строения, в которых длина последовательности звеньев ЭЦА выше, чем у статистических, обладают более высокой термостойкостью (табл. 3). Такой результат становится понятным, если обратиться к механизму образования сополимеров и проанализировать “слабые” связи в макромолекулах [59]. Для сополимеров, синтезированных под действием несимметричного тритиокарбоната ДТМПК (C12H25SC(=S)SC(CH3)2COOH), структуру макромолекул можно представить следующим образом:

В отличие от полимеров, полученных радикальной или анионной полимеризацией, “слабой” связью в макромолекулах, синтезированных методом ОПЦ, является связь C–S между концевым звеном мономера и атомом серы тритиокарбонатного фрагмента [60]. Лабильность связи C–S возрастает при переходе от третичного к четвертичному атому углерода [60]. Следовательно, сополимеры с ω-концевым звеном АН должны быть более термостойкими, чем макромолекулы с ω-концевым звеном ЭЦА.
При единовременной загрузке мономеров ЭЦА расходуется быстро в силу своей высокой реакционной способности, и “голова” макромолекулы будет обогащена звеньями этого мономера (α-конец цепи). В результате звенья акрилонитрила будут с большей вероятностью располагаться в “хвосте” макромолекулы (ω-конец цепи) и соединяться с тритиокарбонатным фрагментом. При непрерывном введении ЭЦА в полимеризацию вероятность его попадания на ω-конец цепи возрастает. Таким образом, градиентные сополимеры действительно должны быть более термостойкими, чем статистические.
Если высказанные выше соображения о частичной термодеструкции, протекающей при 150–250°С, верны, то при изотермической выдержке сополимеров в инертной атмосфере будет происходить уменьшение их ММ. Действительно, нагревание сополимеров при 200 и 225°С приводит к падению ММ сополимеров. Скорость падения ММ растет при повышении температуры (рис. 8) и увеличении доли ЭЦА в сополимере (рис. 9).
Рис. 8.
Нормированные к единичной площади кривые ГПХ сополимера 4 АН и ЭЦА, выдержанные в изотермических условиях при 200 (а) и 225°С (б). Время выдержки: а – 0 (1), 2 (2), 5 (3) и 10 мин (4); б – 0 (1), 1 (2), 2 (3) и 5 мин (4).
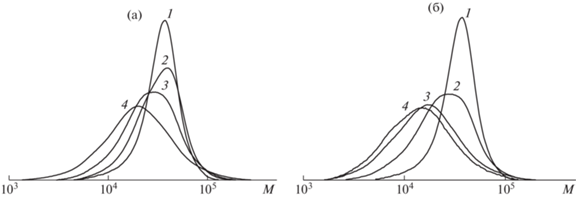
Рис. 9.
Зависимость Mn от времени при изотермической выдержке в инертной атмосфере при 200 (а) и 225°С (б) сополимеров АН и ЭЦА с содержанием ЭЦА 17.0 (4), 11.8 (5) и 4.7 мол. % (6).

Однако частичная термодеструкция не препятствует циклизации нитрильных групп [58]. Для изучения химических превращений, происходящих при нагревании сополимеров, использовали ИК-спектроскопию. Для этого пленки полимеров были прогреты в течение разного времени в инертной атмосфере при 225°С.
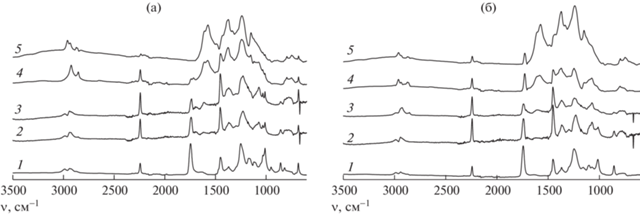
На рис. 10 приведены типичные ИК-спектры сополимеров до и после термообработки. В спектре сополимеров 1 (градиентный) и 5 (статистический) видны характеристические полосы поглощения, отвечающие мономерным звеньям АН и ЭЦА. Валентные колебания нитрильных групп νC≡N обоих мономеров проявляются при 2243 см−1, валентные νСН и деформационные δСНН колебания метиленовых и метиновых групп основной цепи ‒ при 3000–2850 и 1448, 1370 см−1 соответственно. Полоса поглощения валентных колебаний С=О группы ЭЦА видна при 1746 см−1, валентные колебания –С–О– наблюдаются в области 1300–1100 см−1. В ходе циклизации нитрильная группа превращается в иминную –СН=N–. Это приводит к понижению интенсивности полосы поглощения 2243 см−1 (νC≡N), ее уширению и затем к расщеплению на две полосы с максимумами при 2243 см−1 (нитрильная группа в исходном полимере) и при 2200 см−1 (нитрильная группа, участвующая в сопряжении). Одновременно регистрируются полосы поглощения при 1600–1575 см−1, которые соответствуют образованию пиридоновой структуры. Кроме того, в спектрах появляются новые полосы поглощения в области 3000–2700 и 1600–1000 см−1, обусловленные образованием системы сопряженных связей [18]. При этом полоса поглощения при 1746 см−1, отвечающая карбонильной группе νС=О, уменьшается по интенсивности.
Рис. 10.
ИК-спектры, зарегистрированные в ходе термообработки при 225°C сополимеров 1 (а) и 5 (б). Время термообработки 0 (1), 5 (2), 20 (3), 120 (4) и 240 мин (5).
Для сравнительного количественного анализа скорости превращения нитрильных групп по уравнению (1) было рассчитано изменение доли непрореагировавших нитрильных групп φCN от времени (рис. 11). Видно, что в случае градиентных сополимеров (кривые 1 и 2) увеличение доли ЭЦА в цепи вызывает уменьшение скорости превращения общей доли нитрильных групп в сополимере. Аналогичная тенденция характерна и для статистических сополимеров (кривые 4–6).
Рис. 11.
Зависимости общей конверсии нитрильных групп от времени при изотермической обработке в атмосфере аргона при 225°С пленок сополимеров АН и ЭЦА при единовременном (1, 2) непрерывном введении ЭЦА (4–6).

В уравнении (1) не учитывается то обстоятельство, что убыль интенсивности полосы поглощения нитрильных групп обусловлена не только их превращением в иминные группы, но и частичной деструкцией полимера за счет деполимеризации ЭЦА. Тем не менее, если сравнить сополимеры одинакового состава, но разной микроструктуры (1 и 5 или 2 и 4), то видно, что циклизация в градиентных сополимерах протекает быстрее, чем в статистических.
ЗАКЛЮЧЕНИЕ
В работе впервые осуществлена сополимеризация акрилонитрила и этил-2-цианоакрилата по радикальному механизму. Показано, что ЭЦА примерно на три порядка активнее в радикальной сополимеризации, чем АН. Использование агента ОПЦ на основе несимметричного тритиокарбоната (ДТМПК) позволило синтезировать сополимеры с узким ММР и контролируемой ММ. Благодаря большой разнице в активностях мономеров при их единовременной загрузке происходит быстрое расходование ЭЦА и спонтанное образование градиентного сополимера. В результате чего “голова” макромолекулы обогащена звеньями ЭЦА, а “хвост”, соединенный с тритиокарбонатным фрагментом – звеньями АН. Равномерного распределения ЭЦА вдоль цепи можно достичь путем непрерывного введения ЭЦА в сополимеризацию с заданной скоростью. При этом повышается вероятность того, что концевым звеном, соединенным с тритиокарбонатным фрагментом, будет ЭЦА.
Данный аспект определяет различие в термическом поведении градиентных и статистических сополимеров в инертной атмосфере. Градиентные сополимеры обладают более высокой термостойкостью, т.е. для них, по-видимому, циклизация более вероятна, чем деполимеризация. В статистических сополимерах потеря массы происходит при меньших температурах и вклад деполимеризации существеннее. Понижение содержания ЭЦА в сополимерах, независимо от их микроструктуры, приближает их термическое поведение к гомополимеру АН. Так, в статистическом сополимере с 4.6 мол. % ЭЦА превращение нитрильных групп идет примерно с той же скоростью, что и в ПАН.
Таким образом, сополимеры АН и ЭЦА с малым содержанием последнего (до 5 мол. %) позволяют расширить температурный интервал, в котором протекает реакция циклизации, и понизить интенсивность тепловыделения. Это позволяет считать такие сополимеры перспективными объектами для получения ПАН-прекурсоров углеродных волокон.
Список литературы
Chung D.D.L. // Carbon Fiber Composites. Wellin: Butterworth–Heinemann, 1994.
Huang X. // Materials. 2009. V. 2. P. 2369.
Edie D.D. // Carbon. 1998. V. 36. P. 345.
Dalton S., Heatley F., Budd P.M. // Polymer. 1999. V. 40. P. 5531.
Ju A., Guang S., Xu H. // Carbon. 2013. V. 54. P. 323.
Bajaj P., Paliwal D.K., Gupta A.K. // J. Appl. Polym. Sci. 1993. V. 49. P. 823.
Bansal R.C., Donnet J.B. // Compr. Polym. Sci. 1990. V. 6. P. 501.
Bhanu V., Rangarajan P., Wiles K., Bortner M., Sankarpandian M., Godshall D., Wilkes G. // Polymer. 2002. V. 43. P. 4841.
Tsai J.S., Lin C.H. // J. Appl. Polym. Sci. 1991. V. 43. P. 679.
Bajaj P., Roopanwal A.K. // J. Macromol. Sci., Polym. Revs. 1997. V. 37. P. 97.
Rwei S.P., Way T.F., Hsu Y.S. // Polym. Degrad. Stab. 2013. V. 98. P. 2072.
Thomas W.M. // Adv. Polym. Sci. 1961. V. 2. P. 401.
Garcia-Rubio L.H., Hamielec A.E. // J. Appl. Polym. Sci. 1979. V. 23. P. 1397.
Srinivasan N.T., Santappa M. // Makromol. Chem. 1958. V. 26. P. 80.
Izumi Z. // J. Polym. Sci., Polym. Chem. 1967. V. 5. P. 469.
Rahaman M.S.A., Ismail A.F., Mustafa A. // Polym. Degrad. Stab. 2007. V. 92. P. 1421.
Гольдфейн М.Д. // Высокомолек. соед. А. 1990. Т. 32. № 11. С. 2243.
Vashchenko A.F., Toms R.V., Balashov M.S., Pichkunov N., Gervald A.Yu., Prokopov N.I., Maksimov N.M., Plutalova A.V., Chernikova E.V. // Polymer Science B. 2021. V. 63. № 6. P. 802.
Chernikova E.V., Toms R.V., Gervald A.Yu., Proko-pov N.I. // Polymer Science C. 2021. V. 62. № 1. P. 17.
Coover H.W., Dreifus D.W., O’Connor J.T. // Handbook of Adhesives / Ed. by I. Skeist. New York: Van Nostrand Reinhold Co. Inc., 1990. P. 463.
Donnelly E.F., Johnston D.S., Pepper D.C., Dunn D.J. // Polym. Lett. Ed. 1977. V. 15. P. 399.
Pepper D.C., Ryan B. // Makromol. Chem. 1983. V. 184. P. 395.
Pepper D.C., Ryan B. // Makromol. Chem. 1983. V. 184. P. 383.
Eromosele I.C., Pepper D.C. // Makromol. Chem. 1989. V. 190. P. 3095.
Dossi M., Storti G., Moscatelli D. // Macromol. Symp. 2010. V. 289. P. 124.
Pepper D.C. // Makromol. Chem. 1987. V. 188. P. 527.
Johnston D.S., Pepper D.C. // Makromol. Chem. 1981. V. 182. P. 393.
Cronin J.P., Pepper D.C. // Makromol. Chem. 1988. V. 189. P. 85.
Bevington J.C., Jemmett J.A.L., Onyon P.F. // Eur. Polym. J. 1976. V. 12. P. 255.
Bevington J.C., Jemmett J.A.L. // J. Chem. Soc. Faraday Trans. 1 1973. V. 69. P. 1866.
Yamada B., Yoshioka M., Otsu T. // Makromol. Chem. 1983. V. 184. P. 1025.
Yamada B., Hayashi T., Otsu T. // J. Macromol. Sci., Chem. 1983. V. 7. P. 1023.
Yamada B., Kontani T., Yoshioka M., Otsu T. // J. Polym. Sci., Polym. Chem. 1984. V. 22. P. 2381.
Rooney T.R., Mavroudakis E., Lacík I., Hutchinson R.A., Moscatelli D. // Polym. Chem. 2015. V. 6. P. 1594.
Tang H., Tsarevsky N.V. // J. Polym. Sci., Polym. Chem. 2016. V. 54. P. 3683.
Kinsinger J.B., Panchak J.R., Kelso R.L., Bartlett J.S., Graham R.K. // J. Appl. Polym. Sci. 1965. V. 9. P. 429.
Beuermann S., Buback M., Davis T.P., Gilbert R.G., Hutchinson R.A., Olaj O.F., Russell G.T., Schweer J., Van Herk A.M. // Macromol. Chem. Phys. 1997. V. 198. P. 1545.
Полякова А.М., Сучкова М.Д., Магер К.А., Коршак В.В. // Высокомолек. соед. А. 1984. Т. 26. № 1. С. 70.
Hall H.K. Jr., Padias A.B., Chu G., Lee H.Y., Kalinin I., Sansone M., Breckenridge G. // J. Polym. Sci., Polym. Chem. 1992. V. 30. P. 2341.
Kim K.-Y., Park W.-L., Chung Y.-S., Shin D.G., Han J.W. // Carbon Lett. 2011. V. 12. P. 31.
Kim K.-Y., Park W.-L., Shin D.G., Chung Y.-S., Han J.W. // Pat. KR1020110032942A. South Korea. 2011.
Duffy C., Phelan M., Zetterlund P.B., Aldabbagh F. // J. Polym. Sci., Polym. Chem. 2017. V. 55. P. 1397.
Robello D.R., Eldridge T.D., Swanson M.T. // J. Polym. Sci., Polym. Chem. 1999. V. 37. P. 4570.
Birkinshaw C., Pepper D.C. // Polym. Degrad. Stab. 1986. V. 16. P. 241.
Leonard F., Kulkarni R.K., Brandes G., Nelson J., Cameron J.J. // J. Appl. Polym. Sci. 1966. V. 10. P. 259.
Ryan B., McCann G. // Macromol. Rapid Commun. 1996. V. 17. P. 217.
Chorbadjiev K.G., Novakov P.C. // Eur. Polym. J. 1991. V. 27. P. 1009.
Kulkarni R.K., Porter H.J., Leonard F. // J. Appl. Polym. Sci. 1973. V. 17. P. 3509.
Han M.G., Kim S., Liu S.X. // Polym. Degrad. Stab. 2008. V. 93. P. 1243.
Birkinshaw C., Pepper D.C. // Polym. Degrad. Stab. 1986. V. 16. P. 241.
Polymer Handbook/ Ed. by J. Brandrup, E.H. Immergut, E.A. Grulke. New York: Wiley, 1999.
Collins G.L., Thomas N.W., Williams G.E. // Carbon. 1988. V. 26. I. 5. P. 671.
Езриелев А.И., Брохина Э.Л., Роскин Е.С. // Высокомолек. соед. А. 1969. Т. 11. № 8. С. 1670.
Fineman M., Ross S.D. // J. Polym. Sci. 1950. V. 5. № 2. P. 259.
Chiefari J., Chong Y.K., Ercole F., Krstina J., Jeffery J., Le T.P.T., Mayadunne R.T.A., Meijs G.F., Moad C.L., Moad G., Rizzardo E., Thang S.H. // Macromolecules. 1998. V. 31. V. 16. P. 5559.
Chernikova E.V., Mineeva K.O. // Polym. Science C. 2022. V. 64. № 1. P. 1.
Skvortsov I.Y., Varfolomeeva L.A., Kuzin M.S., Vashchenko A.F., Chernikova E.V., Toms R.V., Kulichikhin V.G. // Mendeleev Commun. 2022. V. 32. № 5. P. 652.
Grassie N., McNeill I.C. // J. Polym. Sci. 1958. V. 27. P. 207.
Chernikova E.V., Sivtsov E.V. // Polymer Science B. 2017. V. 59. № 2. P. 117.
Bekanova M.Z., Neumolotov N.K., Jablanovic A.D., Plutalova A.V., Chernikova E.V., Kudryavtsev Y.V. // Polym. Degrad. Stab. 2019. V. 164. P. 18.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)