

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 6, стр. 459-470
КОНТРОЛИРУЕМЫЙ СИНТЕЗ СОПОЛИМЕРОВ N-ВИНИЛАМИДОЯНТАРНОЙ КИСЛОТЫ И ВИНИЛОВОГО СПИРТА ДЛЯ ИММОБИЛИЗАЦИИ НИЗКОМОЛЕКУЛЯРНЫХ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
А. И. Гостев a, Е. В. Сивцов a, *, Д. В. Григорьев b
a Санкт-Петербургский государственный технологический институт (технический университет)
190013 Санкт-Петербург, Московский пр., 24-26/49 лит. А., Россия
b Национальный исследовательский университет ИТМО
197101 Санкт-Петербург, Кронверкский пр., 49, Россия
* E-mail: rheologyspb@gmail.com
Поступила в редакцию 13.10.2022
После доработки 13.11.2022
Принята к публикации 01.12.2022
- EDN: PJYPTP
- DOI: 10.31857/S2308113922700279
Аннотация
Осуществлена контролируемая радикальная полимеризация с обратимой передачей цепи по механизму присоединения‒фрагментации N-винилсукцинимида с винилацетатом в присутствии дибензилтритиокарбоната. Кинетика полимеризации и молекулярно-массовые характеристики полученных полимеров изучены с помощью методов спектроскопии ЯМР и гель-проникающей хроматографии. Проведена оценка значений констант сополимеризации мономеров и описана градиентная микроструктура сополимеров. Получены водорастворимые полимерные матрицы на основе сополимеров N-виниламидоянтарной кислоты и винилового спирта, предназначенные для иммобилизации низкомолекулярных соединений, проявляющих биоактивные свойства. Показано отсутствие бактерицидной активности синтезированных матриц на моделях бактериальных культур. Осуществлен перевод в водорастворимую форму маслорастворимых ремантадина и тримекаина за счет взаимодействия с полученным сополимером.
ВВЕДЕНИЕ
Интерес к созданию полимерных форм лекарственных веществ, использующихся в качестве активных фармацевтических ингредиентов в составе лекарственных средств, не угасает на протяжении нескольких десятилетий. Данное направление выделилось в самостоятельную область на стыке химии высокомолекулярных соединений, медицины и биологии. Обычно идею совмещения уникальных свойств полимеров и низкомолекулярных лекарственных веществ связывают с именами H. Ringsdorf [1, 2] и J. Kopeček [3, 4], относя зарождение целенаправленного использования полимеров для улучшения характеристик лекарственных веществ к 70-м годам XX в. Однако впервые это направление было сформулировано С.Н. Ушаковым на десятилетия ранее [5]. Действенность такого подхода обусловлена пролонгированием действия лекарственных веществ, связанных тем или иным способом с полимерной матрицей, что обеспечивает терапевтическую концентрацию в крови в течение долгого периода времени (до суток), избавляет от передозировок, характерных при приеме низкомолекулярных форм лекарственных веществ, часто снижает токсический эффект и открывает возможности для направленного транспорта лекарственных веществ в определенные органы или ткани [6]. В качестве матриц для создания таких водорастворимых систем известны сополимеры N-виниламидоянтарной кислоты и винилового спирта [7]. Новые синтетические возможности для решения разнообразных задач, в том числе для создания полимеров медико-биологического назначения определенной архитектуры и с заданными молекулярно-массовыми характеристиками, открылись с разработкой методов контролируемой радикальной полимеризации, в частности полимеризации с обратимой передачей цепи по механизму присоединения‒фрагментации (ОПЦ-полимеризация). Механизм, особенности и возникающие на пути реализации трудности ОПЦ-полимеризации в полной мере описаны в обзорах G. Moad, E. Rizzardo и S.H. Thang [8, 9] и двух монографиях [10, 11]. Применительно к сополимерам N-виниламидоянтарной кислоты и винилового спирта, прекурсором которых являются сополимеры N-винилсукцинимида (ВСИ) с винилацетатом (ВА), ОПЦ-полимеризация представляет особый интерес, так как позволяет решить одновременно несколько проблем. Прежде всего это вопрос композиционной неоднородности, характерной для сополимеров, получающихся из разноактивных мономеров, к которым относятся ВСИ и ВА. Другим достоинством является возможность синтеза полимеров с заданной величиной молекулярной массы и узким молекулярно-массовым распределением, что является ключевым требованием для прохождения гистогематического барьера организма [12]. При применении полимеров в составе лекарственных средств их постепенное, но полное выведение из организма обеспечивается ограничением по молекулярной массе, которая не должна превышать нескольких десятков тысяч [7], например 25 × 103 для гиалуроновой кислоты [13], однако пороговые значения могут достигать и (30–50) × 103 в зависимости от гибкости и архитектуры цепи [12]. Кроме того, применение ОПЦ-полимеризации позволяет избежать ряд технологических трудностей. При проведении гомо- и сополимеризации ВСИ в массе мономера кинетика является неконтролируемой, характеризуется ярко выраженным гель-эффектом и часто является причиной получения сшитых, не растворимых ни в одном из растворителей продуктов [14, 15]. Аналогичная проблема существует и в радикальной полимеризации ВА, осложняющаяся еще реакцией передачи цепи на мономер [16, 17]. Эффективный контроль, обеспечиваемый агентом ОПЦ, снижает или даже подавляет гель-эффект, уменьшает роль процессов передачи цепи на полимер, являющихся причиной сшивки [18]. Отдельный вопрос ‒ выбор агента ОПЦ. В настоящей работе в этом качестве использован дибензилтритиокарбонат (БТК), проявивший себя эффективным ОПЦ-агентом в полимеризации широкого ряда винильных мономеров и, в частности, в гомополимеризации ВСИ [19]. Другим важным его достоинством является доступность и относительная простота получения.
Цель настоящей работы – изучение сополимеризации N-винилсукцинимида с винилацетатом в присутствии БТК в условиях обратимой передачи цепи по механизму присоединение‒фрагментация с целью получения на их основе водорастворимых сополимеров N-виниламидоянтарной кислоты с виниловым спиртом, способных к связыванию низкомолекулярных лекарственных веществ, использующихся в качестве активных фармацевтических ингредиентов в составе лекарственных средств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
N-винилсукцинимид синтезировали по методике, описанной в работе [20], очистку проводили путем трехкратной перекристаллизации из раствора в изопропиловом спирте (Tпл = 48.5°С, $n_{D}^{{50}}$ = 1.5020), выход 46%. ЯМР 1H (400.13 Гц, ДМСО-d6): δ = 2.66 (4H, с), 5.02 (1H, д, J = 9.78 Гц), 5.95 (1H, д, J = 16.38 Гц), 6.64 (1H, дд, J = 16.38, 9.78 Гц). Винилацетат перед полимеризацией дважды перегоняли (Tкип = 72°С): ЯМР 1H (400.13 Гц, ДМСО-d6): δ = 2.13 (3H, s), 4.64 (1H, дд, J = 6.32, 1.47 Гц), 4.89 (1H, дд, J = 13.94, 1.47 Гц), 7.21 (1H, дд, J = 13.94, 6.32 Гц). Дибензилтритиокарбонат синтезировали по методике [21]. Выход БТК, представляющего собой кристаллическое вещество желтого цвета, составлял 83.6%. Идентификация по спектру ЯМР 1H (400.13 МГц, 25°C, CDCl3) δ, м.д.: 4.69 (4H CH2, с), 7.28‒7.42 (10H, Ar, м). ДАК дважды перекристаллизовывали из этанола при 50 ± 2°C, и сушили в вакууме при 20°C (Tпл = 104°C). ДМФА и ДМСО (оба – х.ч., Акционерное общество “Вектон”) перед использованием перегоняли в вакууме. NaOH (98%), ч.д.а., KOH (86%), х.ч. (оба – Общество с ограниченной ответственностью “НеваРеактив”), диэтиловый эфир (ч.д.а., Общество с ограниченной ответственностью “Кузбасоргхим”), дейтерированный диметилсульфоксид и дейтерированный хлороформ, 99.5% (оба – Cambridge Isotope Laboratories), LiBr, ≥ 99% (“Sigma Aldrich”, номер продукта 73036), катионит КУ-2-8, H+-форма (Общество с ограниченной ответственностью Производственное объединение “ТОКЕМ”), ремантадина хлоргидрат и тримекаина хлоргидрат (оба – Акционерное общество “Oлайнфарм”), сухой питательный агар (ГРМ-АГАР, Aкционерное общество “ЛенРеактив”, код товара 013057) использовали без дополнительной подготовки. Дистиллированную воду получали на бидистилляторе “Steuergerat purator-bi” (“Kombinat Technisches Glas”, Ilmenau).
Контролируемый синтез сополимеров ВСИ с ВА при содержании ВСИ 25 мол. % в мономерной смеси и разной концентрации агента ОПЦ проводили в массе мономеров при 70°С в течение 72 ч в стеклянных ампулах объемом 10 см3, где создавали инертную атмосферу Ar путем проведения трехкратного цикла замораживание–вакуумирование–размораживание–заполнение Ar. Использовали следующие загрузки: ВСИ 1.50 г, ВА 3.19 г, БТК 0.0025 г (1.8 × 10–3 моль/л), ДАК 0.0007 г (8.6 × 10–4 моль/л), выход сополимера 67%, содержание звеньев ВСИ 34.8 мол. %; ВСИ 1.50 г, ВА 3.22 г, БТК 0.0065 г (4.5 × 10–3 моль/л), ДАК 0.0007 г (8.6 × 10–4 моль/л), выход сополимера 50%, содержание звеньев ВСИ 45.8 мол. %; ВСИ 1.50 г, ВА 3.22 г, БТК 0.0127 г (8.8 × 10–3 моль/л), ДАК 0.0007 г (8.6 × 10–4 моль/л), выход сополимера 35%, содержание звеньев ВСИ 50.6 мол. %. По окончании полимеризации реакционную массу растворяли в 1,4-диоксане и дважды осаждали в воду, сушили в вакууме при 60°С до постоянной массы.
Классическую сополимеризацию ВСИ с ВА в ДМСО ([ДАК] = 2.0 × 10–2 моль/л) при разном составе мономерной смеси проводили в стеклянных ампулах, как описано выше, при 70°С в течение 72 ч. Использовали следующие загрузки: для исходной смеси мономеров состава ВСИ : ВА = = 25 : 75 мол. %, ВСИ 0.161 г, ВА 0.355 г, ДАК 0.0026 г, ДМСО 0.280 г (выход сополимера 41%, содержание звеньев ВСИ 29.0 мол. %); для эквимолярного состава брали ВСИ 0.296 г, ВА 0.209 г, ДАК 0.0033 г, ДМСО 0.5552 г (выход сополимера 51%, содержание звеньев ВСИ 49.3 мол. %); для состава 75 : 25, ВСИ 0.408 г, ВА 0.108 г, ДАК 0.0039 г, ДМСО 0.7625 г (выход сополимера 58%, содержание звеньев ВСИ 59.7 мол. %). По окончании полимеризации сополимер дважды переосаждали из раствора в ДМСО в воду, сушили в вакууме при 80°С до постоянной массы.
Контролируемый синтез сополимеров ВСИ с ВА при разном составе мономерной смеси проводили в стеклянных ампулах, как описано выше, в массе мономеров при 70°С ([ДАК] = 1 × 10–3 моль/л, [БТК] = 5 × 10–3 моль/л) в течение 1, 2.5 и 4 ч. Использовали следующие загрузки: для исходной смеси мономеров состава ВСИ : ВА = 25 : 75 мол. %, ВСИ 2.96 г, ВА 6.12 г, БТК 0.0126 г, ДАК 0.0015 г; для состава 50 : 50, ВСИ 5.92 г, ВА 4.08 г, БТК 0.0145 г, ДАК 0.0018 г; для состава 75 : 25, ВСИ 8.88 г, ВА 2.05 г, БТК 0.0162 г, ДАК 0.0018 г. По окончании полимеризации реакционную массу растворяли в диоксане и дважды осаждали в воду, сушили в вакууме при 60°С до постоянной массы.
Спектры ЯМР 1H регистрировали на спектрометре “Bruker Avance III” с рабочей частотой на ядрах 1Н 400 МГц. Мониторинг сополимеризации ВСИ и ВА проводили при 70°С в специальной ампуле для ЯМР, предназначенной для дегазации/вакуумирования производства WILMAD. В ампуле создавали атмосферу Ar. За начало полимеризации принимали момент через 4 мин после опускания ампулы в нагретую измерительную ячейку. Использовали следующие загрузки: для мономерной смеси, содержащей 50 мол. % ВСИ: ВСИ 0.045 г, ВА 0.031 г, БТК 0.0012 г, ДАК – 0.1 мл раствора ДАК (0.0012 г) в 1 мл ДМСО-d6; для смеси, содержащей 62.5 мол. % ВСИ: ВСИ 0.105 г, ВА 0.043 г, БТК 0.0020 г, ДАК – 0.2 мл раствора ДАК (0.0012 г) в 1 мл ДМСО-d6; для смеси, содержащей 75 мол. % ВСИ: ВСИ 0.131 г, ВА 0.030 г, БТК 0.0020 г, ДАК – 0.2 мл раствора ДАК (0.0012 г) в 1 мл ДМСО-d6.
Для расчета текущих концентраций ВСИ, ВА и БТК производили интегрирование всего спектра от 7.6 до 0 м.д., и этот интеграл нормировали на единичную площадь. Далее по изменению интенсивностей сигналов винильных протонов мономеров и бензильных протонов БТК по отношению к интенсивностям в исходном спектре до начала реакции определяли конверсию мономеров и БТК. Для перехода к мольным концентрациям учитывали изменение объема реакционной смеси в ходе полимеризации. Это делали с использованием коэффициентов контракции мономеров при температуре синтеза. При этом упрощенно предполагали, что объем реакционной смеси является простой суммой объемов ее компонентов (мономеры, передатчик цепи и растворитель – ДМСО-d6, вкладом инициатора пренебрегали). Объем каждого мономера при его конверсии q приближенно описывается линейной функцией
Здесь V0 – объем мономера в исходной смеси, q – его конверсия, k – коэффициент контракции, определяемый на основании значений плотности мономера и полимера (k = (1/ρм ‒ 1/ρп)/(1/ρм)), где ρм и ρп – плотность мономера и полимера соответственно. Были использованы следующие значения k: 0.104 для ВСИ и 0.215 для ВА.Сополимер N-виниламидоянтарной кислоты и винилового спирта получали гидролизом сополимера-прекурсора 5%-ным водным раствором NaOH в течение 48 ч при комнатной температуре. От избытка щелочи сополимер очищали диализом, используя мешок с размером ячейки 1 кДа, затем лиофильно сушили. Полученную солевую форму сополимера N-виниламидоянтарная кислота(Na)‒виниловый спирт переводили в кислотную форму на ионообменной смоле КУ-2-8, взятой в количестве 10%-ного избытка к максимальному расчетному количеству звеньев N-виниламидоянтарной кислоты(Na). Сополимер N-виниламидоянтарной кислоты и винилового спирта подвергали диализу и лиофильно сушили.
Бактерицидную активность сополимеров N-виниламидоянтарной кислоты с N-винилпирролидоном в виде водных растворов концентрацией 0.1, 0.01, 0.001 мг/мг H2O исследовали классическим методом колодцев [22]. Для анализа бактерицидной активности использовали три суточные тест-культуры бактерий: грамотрицательные бактерии Escherichia coli и грамположительные бактерии Staphylococcus aureus и Bacillus cereus. Чистые культуры получали из музея культур кафедры технологии микробиологического синтеза Санкт-Петербургского государственного технологического института (технического университета) и выращивали в течение 1 суток на среде сухого питательного агара при 30°С. Суточные культуры засевали газоном на поверхность среды сухого питательного агара в чашки Петри, после чего в этой агаризованной среде делали колодцы стерильным сверлом. Затем колодцы полностью заполняли исследуемыми растворами полимеров. Опыт повторяли два раза для каждой концентрации сополимеров. Чашки, не переворачивая, помещали в термостат при 30°С на трое суток для роста тест-культур. Анализ бактерицидной активности проводили визуально по наличию (отсутствию) роста тест-культур на поверхности сухого питательного агара вокруг колодцев с исследуемыми сополимерами.
Получение ремантадина-основания и тримекаина-основания проводили из соответствующих хлоргидратов. Хлоргидрат суспендировали в дистиллированной воде и при перемешивании добавляли 1 М NaOH. При этом выделяли активный фармацевтический ингредиент-основание, система расслаивалась на нижний водно-щелочной слой и верхний органический. Целевой продукт экстрагировали диэтиловым эфиром, сушили от следов воды сухим KOH и выделяли отгонкой эфира при вакууме водоструйного насоса. Ремантадин перегоняли в вакууме при 100°С/2 мм. рт. ст. Тримекаин в виде плотного порошка сушили при 20°С/5 мм. рт. ст.
Состав сополимеров ВСИ с ВА определяли на основании данных о содержании в образцах азота. Элементный анализ осуществляли на автоматическом анализаторе марки Vario EL CHNOS Elementar Analyzer (“Elementar Analysensysteme GmbH”, ФРГ). Определяли содержание углерода, водорода, азота и серы. Проводили не менее трех определений для каждого образца. О погрешности определений судили по воспроизводимости результатов, которая составила ±0.1%.
Молекулярно-массовые характеристики сополимеров ВСИ и ВА, полученных полимеризацией в массе мономерной смеси, содержащей 25 мол. % ВСИ, при разной концентрации БТК и сополимеров ВСИ‒ВА, полученных классической сополимеризацией в ДМСО при разном составе мономерной смеси изучали методом ГПХ на хроматографе “Shimadzu” с рефрактометрическим детектированием с применением узкодисперсных полиметилметакрилатных стандартов (диапазон масс (1‒400) × 103). Условия проведения: мобильная фаза – ДМФА с добавлением 0.1% LiBr, 40°C, скорость потока 0.7 мл/мин, предколонка Agilent PLgel 5 μm Guard 50 × 7.5 мм и 2 колонки Agilent PLgel 5 μm MIXED-D, 300 × 7.5 мм, 2‒400 × 103.
ММР сополимеров ВСИ‒ВА, полученных в присутствии БТК при разном составе мономерной смеси, исследовали с помощью высокоэффективной эксклюзионной жидкостной хроматографии на хроматографическом комплексе “Knauer” семейства “Smartline” с рефрактометрическим и спектрофотометрическим детекторами на основе диодной матрицы, с применением узкодисперсных полистирольных стандартов (диапазон масс (1.8‒3100) × 103). Условия проведения: мобильная фаза – 0.5 М раствор LiBr в ДМФА, 30°C, скорость потока 0.3 мл/мин, колонка PL gel MiniMix-B.
ИК-спектры получали на фурье-ИК-спектрометре “Tensor 37” фирмы “Bruker” с помощью приставки НПВО MIRacle фирмы “Pike” с кристаллом ZnSe с алмазным напылением. Спектры регистрировали в диапазоне 4000‒600 см–1 с разрешением 2 см–1 и усреднением по 32 сканированиям.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При полимеризации винильных мономеров в присутствии ОПЦ-агентов, в частности симметричных тритиокарбонатов, контроль над ММ-характеристиками достигается за счет реализации равновесия
 (I)
(I)
В результате происходит постоянный обмен полимерными радикалами Pn, Pm, Pk в макромолекулах, количество которых задается концентрацией агента ОПЦ. Равновесие определяется соотношением констант скоростей реакций присоединения $k_{{ad}}^{{''}}$ макрорадикалов по связи C=S и их отщепления $k_{{fr}}^{{''}}$ от радикального интермедиата. Когда макрорадикалы находятся в свободном состоянии, они, присоединяясь к молекулам мономеров, участвуют в росте цепи. Чем меньшее число новых звеньев успевает присоединиться к радикалу роста до перехода в “спящее” состояние макромолекулы, тем эффективнее контроль. Если не происходит нежелательных побочных реакций, то в результате образуется полимер с узким ММР.
Пара ВСИ и ВА относится к случаю мономеров, сильно различающихся по реакционной способности. В классической радикальной полимеризации это приводит к получению полимеров с ярко выраженной конверсионной композиционной неоднородностью. Ее можно избежать, осуществляя полимеризацию в режиме постепенной загрузки более активного мономера в ходе реакции. Контролируемая радикальная полимеризация предлагает более простой в практическом плане способ избежать такого существенного недостатка. В данном случае состав макромолекул непрерывно меняется вдоль цепи в течение всего времени проведения реакции макромолекул, тогда как между собой они отличаются в наименьшей степени.
Относительная реакционная способность ВСИ и ВА зависит от природы среды, поэтому константы сополимеризации являются константами только для определенных условий проведения процесса (растворитель, концентрация мономеров, инициатор, температура). Так, при сополимеризации в массе с использованием перекиси бензоила в качестве инициатора rВСИ = 5.10, rВА = 0.17 (65°С) [23], rВСИ = 6.05, rВА = 0.18 (60°С) [24], в этаноле с тем же инициатором rВСИ = 5.62, rВА = = 0.17 (70°С), в ДМСО с инициатором ДАК rВСИ = = 2.78, rВА = 0.04 (70°С, ВСИ : ДМСО = 1 : 3), rВСИ = 0.02, rВА = 0.82 (70°С, ВСИ : ДМСО = 1 : 14) [25], в воде с инициирующей системой NH4S2O8‒Na2SO3rВСИ = 1.16, rВА = 0.02 (25°С) [26] и с инициирующей системой трисацетилацетонат марганца‒уксусная кислота rВСИ = 1.13, rВА = 0.30 (25°С) [27]. За исключением случая полимеризации в разбавленном растворе в ДМСО, ВСИ всегда проявляет бóльшую активность.
Для достижения достаточного контроля над молекулярно-массовыми характеристиками приходится учитывать по крайней мере два фактора. С повышением концентрации агента ОПЦ улучшается контроль, но увеличивается вероятность замедления полимеризации (вплоть до полного ингибирования), понижения предельно достижимого выхода полимера и ограничения по величине достижимой ММ. Наоборот, при уменьшении концентрации агента ОПЦ теоретически достижимая степень полимеризации, равная отношению суммарной концентрации мономеров к концентрации агента ОПЦ [ВСИ + ВА]/[БТК], увеличивается, но в ущерб качеству контроля. Гомополимеризация ВА в присутствии БТК сопряжена с рядом трудностей [28], связанных с низким качеством контроля ММ характеристик и ингибированием полимеризации при попытке увеличить концентрацию агента ОПЦ. По отношению к ВСИ дибензилтритиокарбонат, напротив, проявляет себя достаточно эффективным агентом ОПЦ (Ctr = 19) [19]. В связи с этим в присутствии ВСИ следует ожидать удовлетворительного качества контроля, как это наблюдалось при сополимеризации ВА с более активным бутилакрилатом (rВА = 0.01 и rбутилакрилат = 5.38), когда закономерности процесса были близки к гомополимеризации БА [29].
Сополимеризация ВСИ с ВА в присутствии БТК при “неблагоприятном” составе исходной мономерной смеси, содержащей 75 мол. % ВА, приводит к получению полимеров, характеризующихся более узким ММР по сравнению с традиционной радикальной полимеризацией и молекулярной массой, зависящей от концентрации БТК (рис. 1). С повышением концентрации БТК хроматограммы сдвигаются в область меньших ММ, причем только при [БТК] = 8.8 × 10–3 моль/л молекулярно-массовое распределение становится узким и унимодальным.
Рис. 1.
Хроматограммы сополимеров ВСИ с ВА, полученных на предельных конверсиях при 70°С, в присутствии БТК при полимеризации в массе мономерной смеси ВСИ : ВА = 25 : 75, [БТК] × 103 = 1.8 (1), 4.5 (2), 8.8 моль/л (3); [ДАК] = = 8.6 × 10–4 моль/л, а также в условиях классической радикальной полимеризации в ДМСО для смесей ВСИ : ВА = = 25 : 75 (4), 50 : 50 (5) и 75 : 25 мол. % (6).

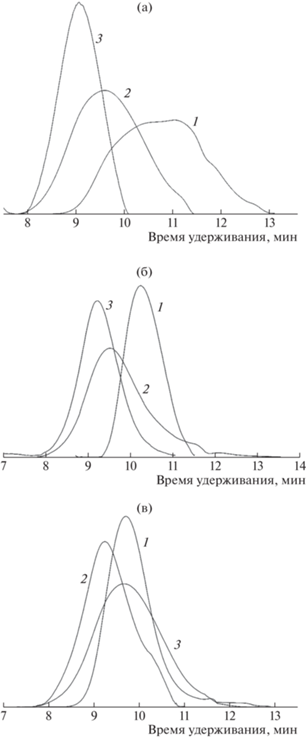
Основным признаком ОПЦ-полимеризации является увеличение ММ с конверсией мономеров. При ОПЦ-сополимеризации ВСИ с ВА в массе хроматограммы образцов, полученных при различной продолжительности полимеризации (табл. 1), сдвигаются в область бóльших значений ММ с увеличением конверсии мономеров (рис. 2), что также подтверждается значениями ММ, полученными из спектров ЯМР 1H полимеров при сравнении интегральных интенсивностей сигналов, относящихся к фенильным протонам концевых бензильных групп и остальным протонам мономерных звеньев. Хроматограммы в основном унимодальны.
Таблица 1.
Характеристики сополимеров N-винилсукцинимида с винилацетатом, полученных в присутствии БТК полимеризацией в массе мономерных смесей различного состава ([БТК] = 5 × 10–3 моль/л, [ДАК] = 1 × 10–3 моль/л, 70°С)
| Состав исходной мономерной смеси ВСИ : ВА, мол. % | Продолжи-тельность полимеризации, ч | Выход сополимера, % | Содержание звеньев ВСИ в сополимере, мол. % | Теоретическая степень полимеризации | Степень полимеризации (по данным спектроскопии ЯМР 1H) |
|---|---|---|---|---|---|
| 25 : 75 | 1.0 | 3.4 | 80 | 75 | 120 |
| 2.5 | 17.5 | 83 | 380 | 450 | |
| 4.0 | 24.4 | 90 | 530 | 500 | |
| 50 : 50 | 1.0 | 4.3 | 82 | 270 | 500 |
| 2.5 | 29.5 | 85 | 560 | 640 | |
| 4.0 | 50.6 | 91 | 960 | 1060 | |
| 75 : 25 | 1.0 | 34.1 | 89 | 580 | 700 |
| 2.5 | 58.2 | 89 | 990 | 1270 | |
| 4.0 | 75.1 | 94 | 1170 | 1560 |
Рис. 2.
Хроматограммы сополимеров ВСИ с ВА, полученных в присутствии БТК полимеризацией в массе мономерной смеси ВСИ : ВА = 25 : 75 (а), 50 : 50 (б), 75 : 25 мол. % (в), 70°С. Продолжительность полимеризации 1 (1), 2.5 (2) и 4 ч (3).
Кинетика сополимеризации ВСИ и ВА изучена методом спектроскопии ЯМР 1H. Понятно, что изучить сополимеризацию в массе мономеров таким способом не представлялось возможным, и мониторинг проводился в растворах концентрацией порядка 10% в ДМСО-d6 (это достаточно точно соответствует соотношению ВСИ : ДМСО = = 1 : 14, для которого определены константы сополимеризации [25]). Следовательно, при интерпретации полученных данных надо учитывать зависимость относительных активностей мономеров от среды. Поскольку интерес представляет поведение системы в условиях эффективного контроля, концентрация БТК была достаточно высокой: 2 × 10–2 моль/л и во всех экспериментах превышала концентрацию ДАК примерно в 25 раз.
С увеличением доли ВА в мономерной смеси брутто скорость сополимеризации и выходы полимера резко снижаются, так что при содержании ВА 50 мол. % достижение сколько-нибудь интересного с практической точки зрения выхода невозможно (рис. 3). Скорость расходования ВСИ и ВА меняется с ходом полимеризации. При содержании ВСИ 62.5 мол. % и более первоначально быстрое вхождение ВСИ в полимерные цепи приводит к снижению его концентрации так, что, начиная с определенного момента, скорость расходования ВА становится выше (рис. 4).
Рис. 3.
Зависимость выхода сополимера от продолжительности реакции при сополимеризации ВСИ и ВА в ДМСО-d6 в присутствии БТК. [ДАК] = 8.0 × × 10–4 моль/л, [БТК] = 2 × 10–2 моль/л, 70°С. Состав исходной мономерной смеси ВСИ : ВА 50 : 50 (1), 62.5 : 37.5 (2) и 75 : 25 мол. % (3).

Рис. 4.
Зависимость конверсии ВСИ (1‒3) и ВА (1'‒3') от продолжительности реакции при сополимеризации ВСИ и ВА в ДМСО-d6 в присутствии БТК. [ДАК] = 8.0 × 10–4 моль/л, [БТК] = 2 × 10–2 моль/л, 70°С. Состав исходной мономерной смеси ВСИ : ВА 50 : 50 (1, 1'), 62.5 : 37.5 (2, 2') и 75 : 25 мол. % (3, 3').

До определенной конверсии происходит обогащение цепей полимера звеньями ВСИ, но после изменения соотношения концентраций мономеров в сторону увеличения доли ВА содержание ВСИ в сополимере незначительно снижается (рис. 5). Скорость расходования БТК можно считать мерой его эффективности как передатчика цепи. В изученном диапазоне составов мономерной смеси БТК расходуется тем быстрее, чем выше содержание ВСИ, но независимо от состава смеси за 6 ч синтеза происходит почти полное исчерпание БТК (рис. 6).
Рис. 5.
Зависимость содержания звеньев ВСИ в сополимере от продолжительности реакции при сополимеризации ВСИ и ВА в ДМСО-d6 в присутствии БТК. [ДАК] = 8.0 × 10–4 моль/л, [БТК] = 2 × 10–2 моль/л, 70°С. Состав исходной мономерной смеси ВСИ : ВА 50 : 50 (1), 62.5 : 37.5 (2) и 75 : 25 мол. % (3).
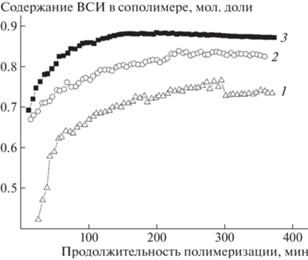
Рис. 6.
Зависимость конверсии БТК от продолжительности реакции при сополимеризации ВСИ и ВА в ДМСО-d6 в присутствии БТК. [ДАК] = 8.0 × 10–4 моль/л, [БТК] = 2 × 10–2 моль/л, 70°С. Состав исходной мономерной смеси ВСИ : ВА 50 : 50 (1), 62.5 : 37.5 (2) и 75 : 25 мол. % (3).

На основании данных об изменении состава сополимера с ходом реакции при разном составе мономерной смеси возможна оценка констант сополимеризации. Методами Езриелева–Брохиной–Роскина [30] и Келена–Тюдоша [31, 32] были рассчитаны константы сополимеризации винилацетата и N-винилсукцинимида, которые составили: rВА < 0, rВСИ = 0.77 ± 0.09 (метод Езриелева–Брохиной–Роскина) и rВA < 0, rВСИ = 0.79 ± ± 0.09 (метод Келена–Тюдоша) при учете данных, полученных в диапазоне выхода сополимера до 10% и rВА < 0, rВСИ = 1.23 ± 0.17 (метод Езриелева–Брохиной–Роскина) и rВA < 0, rВСИ = 1.27 ± 0.24 (метод Келена–Тюдоша) при учете данных, полученных в диапазоне выходов сополимера до 35% (метод Келена–Тюдоша может применяться с достаточной точностью с использованием данных, полученных при конверсиях мономеров до 50% [32]). Отрицательное значение константы сополимеризации лишено физического смысла, однако может быть получено при формальном применении алгоритмов расчета. Такой результат можно интерпретировать как значение константы, близкое к нулю. Интересно, что в данном эксперименте, отличающемся от описанного ранее [25] тем, что в качестве растворителя использовали дейтерированный ДМСО, не наблюдалось инверсии активностей мономеров, и ВСИ проявлял заметно более высокую реакционную способность.
Знание констант сополимеризации предоставляет возможность оценки микроструктуры цепи сополимера. Для вероятностей образования диад M1‒M1 (f11), M1‒M2 (f12), M2‒M1 (f21) и M2‒M2 (f22) могут использоваться уравнения [33]
(2)
${{f}_{{11}}} = \frac{{{{r}_{1}}{{{\left( {\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}}} \right)}}^{2}}}}{{{{r}_{1}}{{{\left( {\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}}} \right)}}^{2}} + 2\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}} + {{r}_{2}}}},$(3)
${{f}_{{12}}} = {{f}_{{21}}} = \frac{{\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}}}}{{{{r}_{1}}{{{\left( {\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}}} \right)}}^{2}} + 2\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}} + {{r}_{2}}}},$(4)
${{f}_{{22}}} = \frac{{{{r}_{2}}}}{{{{r}_{1}}{{{\left( {\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}}} \right)}}^{2}} + 2\frac{{\left[ {{{{\text{M}}}_{1}}} \right]}}{{\left[ {{{{\text{M}}}_{2}}} \right]}} + {{r}_{2}}}}$На основе этих уравнений были выведены функции распределения вероятностей образования структур, состоящих из n последовательно соединенных звеньев каждого типа F1n для мономера M1 и F2n для мономера M2 [34]:
(5)
${{F}_{{1n}}} = {{f}_{{21}}}{{\left( {\frac{{{{f}_{{11}}}}}{{{{f}_{{11}}} + {{f}_{{12}}}}}} \right)}^{{n - 1}}}\frac{{{{f}_{{12}}}}}{{{{f}_{{11}}} + {{f}_{{12}}}}}n,$(6)
${{F}_{{2n}}} = {{f}_{{12}}}{{\left( {\frac{{{{f}_{{22}}}}}{{{{f}_{{22}}} + {{f}_{{21}}}}}} \right)}^{{n - 1}}}\frac{{{{f}_{{21}}}}}{{{{f}_{{22}}} + {{f}_{{21}}}}}n.$Зная f11, f12, f21 и f22, можно вычислить среднюю длину блоков последовательно соединенных звеньев каждого типа [34]:
Данные параметры микроструктуры были определены для сополимеров ВA c ВСИ, исходя из значений констант сополимеризации rВA = 0.01, rВСИ = 0.79. Замена отрицательного значения rВA на любое число, близкое к нулю, приводит к одинаковым результатам для f11, f12(f21), f22, F1n, F2n, $\overline {{{L}_{1}}} $ и $\overline {{{L}_{2}}} ~$ (табл. 2).
Таблица 2.
Основные параметры микроструктуры цепи сополимеров винилацетата и N-винилсукцинимида в зависимости от состава мономерной смеси
| Вид структуры | Содержание винилацетата (M1) в мономерной смеси, мол. % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | |
| f11 | 0 | 0 | 0.0001 | 0.0002 | 0.0004 | 0.0006 | 0.001 | 0.0018 | 0.0043 |
| f12 = f21 | 0.110 | 0.194 | 0.260 | 0.314 | 0.358 | 0.396 | 0.427 | 0.454 | 0.477 |
| f22 | 0.780 | 0.612 | 0.480 | 0.372 | 0.283 | 0.208 | 0.145 | 0.090 | 0.0420 |
| $\overline {{{L}_{1}}} $ | 1.000 | 1.000 | 1.000 | 1.001 | 1.001 | 1.002 | 1.002 | 1.004 | 1.009 |
| $\overline {{{L}_{2}}} ~$ | 8.11 | 4.16 | 2.84 | 2.18 | 1.79 | 1.53 | 1.34 | 1.20 | 1.09 |
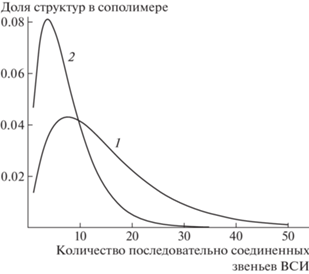
При интерпретации данных (табл. 2), полученных при высоком содержании ВСИ в мономерной смеси, не следует считать, что, например, при содержании ВСИ 90 мол. % в сополимере блоки преимущественно из 8 звеньев ВСИ чередуются с индивидуальными звеньями ВА. Распределение вероятностей образования последовательностей F1n, F2n широкое (рис. 7), и доля длинных блоков, содержащих десятки звеньев ВСИ, может быть существенной.
Рис. 7.
Зависимость доли последовательностей звеньев ВСИ в цепи сополимера ВСИ с ВА от их длины. Содержание ВСИ в смеси 90 (1) и 80 мол. % (2).
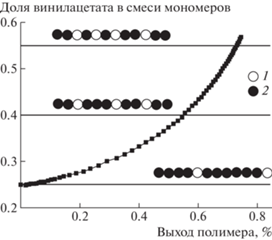
Поскольку в присутствии достаточного количества БТК сополимеризация ВСИ с ВА проходит в режиме “живых” цепей, результатом является получение градиентных сополимеров. Зная, как меняется состав мономерной смеси в ходе реакции, можно оценить изменение микроструктуры по длине цепи. Например, при сополимеризации мономерной смеси, содержащей 25 мол. % ВА, за 6 ч синтеза его доля возрастает до 57 мол. % благодаря быстрому расходованию ВСИ, что приводит к сокращению длины блоков ВСИ и увеличению содержания ВА на участках цепи, полученных на поздних стадиях процесса (рис. 8).
Рис. 8.
Изменение соотношения мономеров при полимеризации смеси, содержащей 25 мол. % ВА с выходом полимера. Схематически изображена микроструктура цепи, соответствующая содержанию ВА 25, 40 и 55 мол. % в мономерной смеси. 1 – звенья ВА, 2 – звенья ВСИ.
При комнатной температуре сополимеры ВСИ с ВА легко подвергаются щелочному гидролизу, давая сополимеры N-виниламидоянтарной кислоты и винилового спирта после перевода полученных полимеров в кислотную форму:
 (II)
(II)
Протекание гидролиза и дальнейший переход сополимера из солевой формы в кислотную подтвержден анализом ИК-спектров сополимеров ВСИ‒ВА, N-виниламидоянтарная кислота(Na)‒ виниловый спирт и N-виниламидоянтарная кислота‒виниловый спирт (рис. 9). В спектре исходного сополимера ВСИ‒ВА имеются полосы поглощения, соответствующие симметричным νC=O = 1768 см–1 (плечо на интенсивной полосе 1731 см–1) и асимметричным νC=O = 1693 см–1 валентным колебаниям связей C=O в сукцинимидном цикле, валентным колебаниям связи C=O винилацетатного звена νC=O = 1731 см–1, валентным колебаниям C–N-связи νC–N = 1369 см–1, валентным колебаниям связи C–O в составе сложноэфирной группы νC–O = 1229 см–1. После гидролиза в спектре сополимера N-виниламидоянтарная кислота(Na)‒виниловый спирт появляются характеристические полосы валентных колебаний OH-связей νOH = 3267 см–1 и NH-связи νNH = 3082 см–1 (широкие сигналы в области 3000‒3600 см–1). После раскрытия сукцинимидного цикла в спектре наблюдаются валентные колебания связи C=O в составе амидного фрагмента νC=O = 1637 см–1 и в составе карбоксильной группы νC=O = 1562 см–1. Интенсивная полоса δNCH = 1403 см–1 относится к деформационным колебаниям амидного фрагмента. При переводе сополимера в кислотную форму положение полосы валентных колебаний связи C=O в составе амидного фрагмента практически не меняется νC=O = 1629 см–1, а полоса валентных колебаний связи C=O карбоксильной группы сдвигается в область бóльших волновых чисел νC=O = 1714 см–1. Полосы валентных колебаний групп OH и связи NH сохраняются в области 3000–3600 см–1, но у полосы νOH = 3278 см–1 появляется плечо около 3400 см–1, что является следствием наложения полос, относящихся к OH карбоксильной группы и OH звена винилового спирта. Интенсивная полоса деформационных колебаний амидного фрагмента δNCH = 1405 см–1 также практически не меняет своего положения.
Рис. 9.
ИК-спектры сополимеров ВСИ‒ВА (1), N-виниламидоянтарная кислота (Na)‒виниловый спирт (2) и N-виниламидоянтарная кислота‒виниловый спирт (3).

Для подтверждения соответствия полученных матриц требованиям, предъявляемым к медицинским полимерам, было проведено исследование их биологической активности в средах, содержащих бактериальные культуры Escherichia coli, Staphylococcus aureus, Bacillus cereus. Вокруг колодцев, заполненных растворами концентрации 0.001–0.1 мг/мг H2O сополимеров N-виниламидоянтарной кислоты с виниловым спиртом, содержащих 35–51 мол. % звеньев N-виниламидоянтарной кислоты, не наблюдали зон отсутствия роста тест-культур бактерий, что свидетельствует об отсутствии биологической активности сополимера по отношению к исследованным типам бактерий. А так как рассматриваемые образцы оказались биологически пассивными в отношении Escherichia-coli – бактерий нормальной микробиоты кишечника человека, с большой вероятностью они не окажут отрицательного воздействия и на полезную микрофлору кишечника человека.
Способность полученных водорастворимых полимеров N-виниламидоянтарной кислоты и винилового спирта к иммобилизации низкомолекулярных лекарственных веществ-оснований изучена на примере ремантадина и тримекаина, имеющих в своем составе первичную и третичную аминогруппы соответственно. Для иммобилизации использовали образцы, полученные гидролизом сополимеров ВСИ‒ВА, синтезированных в массе в присутствии БТК из мономерной смеси ВСИ : ВА = 25 : 75, хроматограммы которых приведены на рис. 1. Сополимеры содержали 35, 46 и 51 мол. % звеньев N-виниламидоянтарной кислоты, что является разумным для количества якорных групп. Оба лекарственных вещества не растворяются в воде, но при совмещении их с водными растворами сополимеров N-виниламидоянтарной кислоты и винилового спирта с концентрацией 0.01 осново-моль/л, взятых из расчета 10%-ного мольного избытка звеньев N-виниламидоянтарной кислоты по отношению к аминогруппам лекарственного вещества, происходит их растворение и переход в раствор, что связано с образованием ионной связи между лекарственным веществом-основанием и полимерной матрицей:
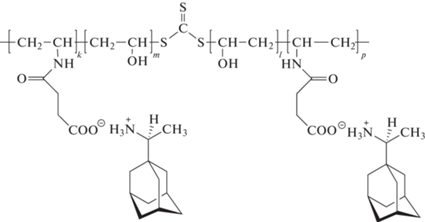 (III)
(III)
 (IV)
(IV)
ЗАКЛЮЧЕНИЕ
Проведение сополимеризации N-винилсукцинимида с винилацетатом в условиях обратимой передачи цепи по механизму присоединения-фрагментации в присутствии дибензилтритиокарбоната позволяет получить композиционно однородные узкодисперсные сополимеры с контролируемой молекулярной массой, удовлетворяющей требованиям, которые предъявляются к полимерам, применяемым в составе лекарственных средств. N-винилсукцинимид характеризуется бóльшей активностью, чем винилацетат, как при сополимеризации в массе мономеров, так и в растворе в дейтерированном диметилсульфоксиде.
Водорастворимые сополимеры N-виниламидоянтарной кислоты и винилового спирта, не проявляющие бактерицидной активности на моделях бактериальных культур Staphylococcus aureus, Bacillus cereus, а также Escherichia-coli – родственной нормальной микрофлоре кишечника, получают щелочным гидролизом сополимеров N-винилсукцинимида с винилацетатом с последующим переводом в кислотную форму. На их основе могут быть получены водорастворимые формы маслорастворимых активных фармацевтических ингредиентов, например ремантадина и тримекаина.
Авторы выражают благодарность С.Г. Изотовой СПбГТИ(ТУ), за анализ данных ИК-спектроскопии.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации (Госзадание 0785.00.Х6019).
Список литературы
Ringsdorf H. // J. Polym. Sci., Polym. Symp. 1975. V. 51. № 1. P. 135.
Bader H., Ringsdorf H., Schmidt B. // Angew. Makromol. Chem. 1984. V. 123. № 1. P. 457.
Kopeček J. // Polim. Med. 1977. V. 7. № 3. P. 191.
Yang J., Kopeček J. // Encyclopedia of Polymeric Nanomaterials/ Ed. by S. Kobayashi, K. Müllen. Berlin; Heidelberg: Springer, 2014.
Ушаков С.Н. Синтетические полимеры лекарственного назначения. Л.: Медгиз, 1962.
Duncan R. // J. Controll. Release. 2014. V. 190. P. 371.
Шальнова Л.И., Лавров Н.А., Сельков С.А., Платонов В.Г., Зубрицкая Н.Г., Иванова Т.В., Машина Л.С. // Изв. СПбГТИ(ТУ). 2013. Т. 19(43). С. 55.
Moad G., Rizzardo E., Thang S.H. // Aust. J. Chem. 2005. V. 58. № 6. P. 379.
Moad G., Rizzardo E., Thang S.H. // Aust. J. Chem. 2012. V. 65. № 8. P. 985.
Handbook of RAFT Polymerization / Ed. by C. Barner-Kowollik. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2008.
RAFT Polymerization: Methods, Synthesis, and Applications / Ed. G. Moad, E. Rizzardo. Wiley-VCH GmbH, 2021.
Duncan R. // Nat. Rev. Drug Discovery. 2003. V. 2. P. 347.
Fraser J.R.E., Laurent T.C., Pertoft H., Baxter E. // Biochem. J. 1981. V. 200. P. 415.
Nikolaev A.F., Ushakov S.N., Krasnosel’skaya I.G. // Russ. Chem. Bull. 1959. V. 8. № 9. P. 1564.
Даниэль Н.В., Николаев А.Ф. // Высокомолек. соед. 1966. Т. 8. № 3. С. 465.
Polymer Handbook / Ed. by J. Brandrup, E.H. Immergut, E.A. Crulue. New York: Wiley, 1999.
Nozakura S.-I., Morishima Y., Murahashi S. // J. Polym. Sci., Polym. Chem. Ed. 1972. V. 10. № 10. P. 2781.
Chernikova E.V., Sivtsov E.V. // Polymer Science B. 2017. V. 59. № 2. P. 117.
Sivtsov E.V., Gostev A.I., Parilova E.V., Dobrodumov A.V., Chernikova E.V. // Polymer Science C. 2015. V. 57. № 1. P. 110.
Levit M., Vdovchenko A., Dzhuzha A., Zashikhina N., Katernyuk E., Gostev A., Sivtsov E., Lavrentieva A., Tennikova T., Korzhikova-Vlakh E. // Int. J. Mol. Sci. 2021. V. 22 (21). № 11457.
Pat. 6369158B1 USA. 2002.
Куличенко Е.О., Андреева О.А., Лукашук С.П., Мазурина М.В. // Фармация и фармакология. 2015. № 4 (11). С. 4.
Николаев А.Ф., Ушаков С.Н., Мишкилеева Л.С. // Высокомолек. соед. 1964. Т. 6. № 2. С. 287.
Furukawa J., Tsuruta T., Fukutani H., Yamamoto N., Shiga M. // J. Soc. Chem. Industry, Jpn. 1957. V. 60. № 3. P. 353.
Бондаренко С.Г., Николаев А.Ф., Баранова С.А., Пляшечник Н.И., Смирнова Г.А., Обухова С.В., Байденок И.В., Степанов Е.М., Глущенок И.Н., Андреева Е.Д. // Высокомолек. соед. А. 1981. Т. 23. № 12. С. 2639.
Лавров Н.А., Николаев А.Ф., Лепшина Е.М., Лаврова Т.В. // Журн. прикл. хим. 1992. Т. 65. № 9. С. 2111.
Лавров Н.А. // Журн. прикл. хим. 1994. Т. 67. № 9. С. 1547.
Chernikova E.V., Yulusov V.V., Mineeva K.O., Golubev V.B., Garina E.S. // Polymer Science B. 2011. V. 53. N. 7–8. P. 437.
Chernikova E.V., Yulusov V.V., Mineeva K.O., Garina E.S., Sivtsov E.V. // Polymer Science B. 2012. V. 54. № 7–8. C. 349.
Ezrielev A.I., Brokhina E.L., Roskin E.S. // Polym. Sci. A. 1969. V. 11. № 8. P. 1670.
Tüdõs F., Kelen T., Földes-Berezhnykh T., Turcsányi B. // React. Kinet. Catal. Letter. 1975. V. 2. № 4. P. 439.
Rao S.P., Ponratnam S., Kapur S.L., Iyer P.K. // J. Polym. Sci., Polym. Lett. Ed. 1976. V. 14. № 9. P. 513.
Гиндин Л.М., Абкин А.Д., Медведев С.С. // Докл. АН СССР. 1947. Т. 56. № 2. С. 177.
Wall F.T. // J. Am. Chem. Soc. V. 66. № 12. P. 2050.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)