

Химия высоких энергий, 2020, T. 54, № 4, стр. 277-282
Изучение переноса заряда в радикальных реакциях отрыва атома водорода с участием триплетных нитросоединений
Д. А. Фомичев a, *, Д. В. Овсянников a, С. В. Зеленцов a
a ФГАОУВО “Нижегородский государственный университет им. Н.И. Лобачевского”
603950 Нижний Новгород, просп. Гагарина, 23, Россия
* E-mail: xomachiner@gmail.com
Поступила в редакцию 13.09.2019
После доработки 20.02.2020
Принята к публикации 28.02.2020
Аннотация
Проверена гипотеза об образовании анион-радикала в ходе темновой стадии реакции переноса атома водорода от молекулы сероводорода к молекуле триплетного нитросоединения методами квантовой химии. Анализ изменения спиновой плотности в процессе реакции, как и анализ изменений электронной плотности в реальном времени с помощью теории функционала плотности (методом RT TD-DFT) не выявил переноса заряда между реагентами. В то же время, обнаруженная слабая зависимость энергетических параметров реакции от диэлектрической проницаемости растворителя свидетельствует в пользу гипотезы о переносе заряда.
ВВЕДЕНИЕ
Разработка низкотемпературных способов окисления органических соединений, в том числе содержащих атомы серы, считается весьма актуальной задачей. Заманчивым ее решением является использование фотохимических реакций для окисления серосодержащих соединений [11]. Гипотетически, использование фотохимической реакции должно снизить необходимое количество энергии для проведения окисления, а также отказаться от использования катализаторов, содержащих тяжелые металлы. В химической литературе неоднократно появлялись сообщения о возможности использования нитросоединений в качестве фотохимических окислителей [5, 7, 10]. Помимо основной задачи окисления органического субстрата, подобный подход позволяет решить проблему утилизации нитросоединений, огромные количества которых подлежат переработке в химической промышленности (например, образующиеся в качестве побочных продуктов в производстве анилиновых красителей). Нитросоединения обладают крайне высоким квантовым выходом интеркомбинационной конверсии [30], что делает их перспективными кандидатами на роль окислителей в данном процессе. К сожалению, выход продуктов реакции окисления значительно ниже, чем ожидается в связи со столь высоким выходом триплетного нитросоединения [29]. По этой причине возникает необходимость дальнейшего изучения механизма фотовосстановления триплетного нитросоединения, с целью выявления причин уменьшения выхода продуктов окисления.
Механизмы фотовосстановления ароматических нитросоединений изучались экпериментально [9, 10, 23] и теоретически [16, 28]. В частности, в [2] на основе полученных данных была выдвинута гипотеза о том, что переносу атома водорода от молекулы амина к молекуле нитронафталина предшествует стадия переноса электрона с образованием анион-радикала нитронафталина и катиона амина. В последствии происходит перенос катиона водорода, выравнивающий заряд, а также приводящий к образованию радикального центра на атоме азота амина.
Без широкого изучения таких реакций разработка способов увеличения их эффективности, по-видимому, невозможно.
Целью данной работы является выявление переноса заряда между молекулой нитросоединения и восстановителем в процессе темновой стадии фотохимического окисления сероводорода нитрометаном. Для достижения данной цели проведено моделирование указанной реакции с участием молекулы нитросоединения в триплетном состоянии, включающее в себя анализ спиновой плотности и изучение протекания этой реакции в различных по величине полярности растворителях. Кроме того, для получения полной картины изменения заряда в процессе реакции проведен анализ электронной плотности в реальном времени с помощью метода RT TD-DFT.
МЕТОДИЧЕСКАЯ ЧАСТЬ
Для проведения квантово-химических расчетов в рамках данной работы использовался программный пакет North-West Chemistry версии 6.6 [24]. Для расчeта энергетических параметров реакции, а также распределения спиновой плотности в молекулах реагентов и продуктов использована теория функционала плотности в сочетании с функционалом PBE0 и базисным набором 6-311++G(d, p). Выбор уровня теории и использованного базиса сделан, исходя из краткого методологического исследования, проведенного в работе [6].
Анализ спиновой плотности в процессе переноса атома водорода осуществлен по следующей методике. Первым шагом было нахождение оптимальных геометрических параметров исходного и результирующего состояния системы нитрометан–сероводород. Затем с помощью процедуры Nudged Elastic Band (NEB) [12, 13] программного пакета NWChem были определены геометрические параметры участников реакции в промежуточных точках реакционного пути, включающих переходное состояние. Для каждой точки на полученном профиле ППЭ было найдено распределение спиновой плотности. Принимая во внимание характер изменения спиновой плотности, был сделан вывод о наличии или отсутствии переноса заряда между молекулами исходных веществ, перед достижением системой переходного состояния.
Анализ электронной плотности в реальном времени проведен методом RT TD-DFT [17] с использованием того же базисного набора и функционала, который был использован при анализе спиновой плотности. Моделирование переноса заряда проводилoсь следующим образом. Сначала, с помощью метода TD-DFT были получены оптимизированные геометрические параметры, а также данные о молекулярных орбиталях для изолированных молекул сероводорода и нитрометана. Кроме того, тем же методом были получены оптимизированные геометрические параметры предреакционного состояния системы нитрометан–сероводород. В дальнейшем, данные о молекулярных орбиталях изолированных молекул были использованы вместе с геометрическими параметрам предреакционного состояния системы в качестве отправной точки при моделировании перераспределения электронной плотности методом RT TD-DFT. Предполагается, что распределение спиновой плотности в разделенных молекулах и предреакционном комплексе не будет соответствовать друг другу, и, таким образом, по результатам расчета можно будет судить о перераспределении электронной плотности, т.е. о переносе заряда между реагентами [21]. Методом RT TD-DFT проводилось моделирование 500 нс изменений электронной плотности молекул реагентов, затем была проанализирована зависимость изменения заряда на атомах реагентов от времени. Для оценки влияния диэлектрической константы растворителя на энергетические параметры реакции была использована модель растворителя COSMO [14, 18, 27]. Данные о растворителях взяты из литературы [25]. Энергии активации и энтальпии реакций получены методом DFT PBE0/6-311++G** с учетом энергии нулевых колебаний.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Процесс переноса электрона между реагентами в реакции отрыва атома водорода можно проследить по изменению спиновой плотности в реакционной системе. Перенос электрона от молекулы сероводорода повлечет за собой появление на ней спиновой плотности, поскольку число электронов молекулы станет нечетным. В свою очередь, превращение бирадикала нитросоединения в триплетном состоянии в анион-радикал будет сопровождаться уменьшением спиновой плотности на одном из атомов кислорода нитрогруппы за счет образования электронной пары с электроном, перенесенным от молекулы сероводорода.
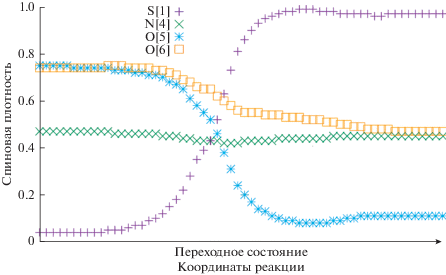
Анализ изменения спиновой плотности при движении системы по профилю ППЭ, соответствующему реакции переноса атома водорода, показал, что перенос электрона осуществляется одновременно с переносом протона, а не предшествует ему. Из рис. 2 видно, что спиновая плотность на пятом атоме кислорода (согласно нумерации на рис. 1) уменьшается синхронно с изменением координаты реакции и при достижении системой переходного состояния снижается до 0.55. Это означает, что гипотеза об образовании анион-радикала в первом элементарном акте темновой стадии реакции не подтверждается, т.к. заряд переносимого электрона (перемещение электрона можно отследить по изменению спиновой плотности) остается скомпенсированным зарядом переносимого протона (движение протона соответствует изменению координаты реакции). Фактически, речь идет о переносе нейтрального атома водорода, не сопровождающегося переносом заряда в процессе превращения. Для дальнейшей проверки гипотезы были рассмотрены результаты моделирования электронной плотности в реагентах.
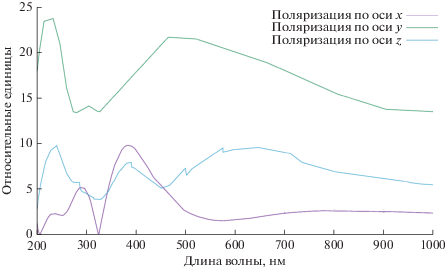
Изучение поведения электронной плотности методом RT TD-DFT показало отсутствие каких-либо осцилляций заряда между молекулами сероводорода и нитрометана на протяжении всего времени моделирования. Однако были обнаружены осцилляции электронной плотности, не связанные с межмолекулярным переносом заряда (рис. 3). В спектре осцилляций можно выделить несколько пиков: в районе 240, 290, 390, 500 и 640 нм. Данные пики сложно отнести к спектру любого из реагентов, что может являться косвенным свидетельством образования предреакционного комплекса.
Рис. 3.
Спектр осцилляций электронной плотности в системе нитрометан–сероводород, обнаруженных методом RT TD-DFT.
В случае образования анион-радикала, следует ожидать сильную зависимость энергетических параметров реакции от полярности растворителя [15] поскольку более полярные растворители стабилизируют анион-радикал. На рис. 1 приведен график зависимости энтальпии и энергии активации реакции от диэлектрической константы растворителя.
Из рис. 4 следует, что изменение полярности растворителя приводит к изменению энергии активации реакции не более, чем на 3 ккал/моль. Зависимость энергии активации от диэлектрической константы слабо соответствует линейной (R2 = 0.62), причем наибольшее отклонение имеют точки, связанные с такими растворителями как ДМСО и муравьиная кислота. Резкое падение энергии активации в случае муравьиной кислоты вероятно связанно с тем, что муравьиная кислота, как и вода является протонным растворителем с высокой способностью к образованию водородных связей [8].
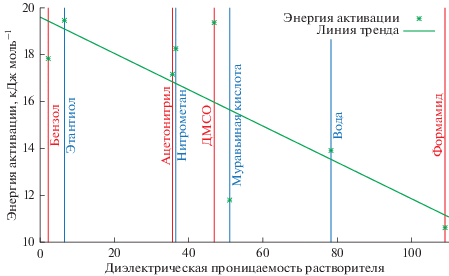
При исключении точек, соответствующих ДМСО и муравьиной кислоте зависимость более соответствует с линейной (R2 = 0.93).
Полученный результат мог бы свидетельствовать в пользу образования анион-радикала, однако для этого необходимо найти объяснение выпадению энергетических параметров реакции в ДМСО и муравьиной кислоте из общего тренда.
На рис. 5 можно заметить тренд синхронного изменения энергии активации и энтальпии. Между изменением энтальпии и энергии активации действительно есть линейная корреляция (R2 = 0.97). Такая же корреляция наблюдается для реакций радикального присоединения [3]. Это позволяет предположить общий характер зависимости между энергией активации и энтальпией для реакции радикального отрыва атома водорода и радикального присоединения.
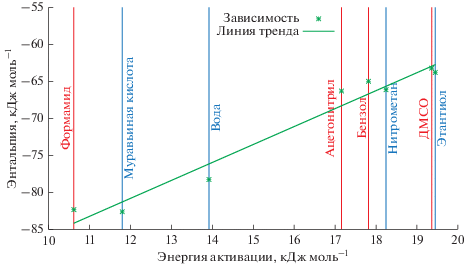
Полученные результаты моделирования не вполне согласуются с данными полученными для аналогичных реакций с участием аминов в качестве доноров атома водорода [2, 4, 9]. Для аминов имеются различные доказательства переноса заряда, в то время как в данной работе перенос заряда обнаружить не удалось. Самой вероятной причиной расхождений может быть отличие в механизмах реакции нитросоединений с соединениями двухвалентной серы от реакций с аминами. В частности, для аминов предложен механизм реакции, в ходе которого перенос заряда осуществляется от синглетного нитросоединения к амину и только после этого происходит формирование предреакционного комплекса и его возбуждение в триплетное состояние [1]. О наличии предреакционного комплекса в случае аминов также свидетельствуют результаты квантово-химического моделирования [20]. Кроме того, в условиях эксперимента происходит гораздо большее число одновременных параллельных реакций, в то время как моделирование было проведено лишь для одной из стадий. В работе [26] отмечается, что в процессе образования анион-радикала активное участие принимает растворитель, играя роль донора электрона. В частности, большую роль играет присутствие соляной кислоты в системе [2]. Однако, в рамках выбранной модели перенос электрона с молекул растворителя невозможен.
Перенос электрона подтверждается для систем с аминами в качестве восстановителей [4], однако для серосодержащих соединений подобных подтверждений найти не удалось. Еще одним отличием моделируемой системы от известных случаев переноса электрона является расположение уровней в молекуле окислителя: в нитронафталинах, для которых были найдены аргументы в пользу переноса электрона, нижним триплетным состоянием является (π, π*), в то время как для моделируемого нитрометана – (n, π*) [19, 22]. Таким образом, полученные данные могут свидетельствовать об образовании анион-радикала, но на более ранних реакционных стадиях.
ЗАКЛЮЧЕНИЕ
Нами не выявлено убедительных признаков переноса электрона, предшедствующего переносу протона на темновой стадии реакции радикального переноса атома водорода от молекулы сероводорода к триплетному бирадикалу нитрометана. Против гипотезы об образовании анион-радикала свидетельствует динамика изменения спиновой плотности и отсутствие осциляций заряда при моделировании системы методом RT TD-DFT. Однако моделирование реакции в различных растворителях показало слабую зависимость энергетических параметров от диэлектрической константы растворителя, что может рассмaтриваться как косвенный признак образования анион-радикала.
Список литературы
Зеленцов С.В. и др. // Химия высоких энергий. 2004. № 2(38). С. 142.
Cu A., Testa A.C. // J. American Chemical Society. 1974. № 6(96). C. 1963.
Denisov E.T. // Russian Chemical Reviews. 2000. № 2(69). C. 153.
Dopp D., Muller D., Sailer K.H. // Tetrahedron Letters 1974. № 24. C. 2137–2140.
Fawi M., Latif A.E. // J. Photoch. Photobiol. A: Chemistry. 2001. № 2–3(141). C. 241.
Fomichev D., Plechovich S., Zelentsov S. // MDPI. 2013. e007 c.
Frolov A.N., Kuznetsova N.A., El’tsov A.V. // Rus. Chem. Rev. 1976. № 11(45). C. 1024.
Gómez S. et al. // Int. J. Quan. Chem. 2019. № 4(119). C. e25814.
Görner H., Döpp D. // J. Chem. Soc., Perkin Transactions 2. 2002. № 1. C. 120.
Gruen H., Görner H. // Photochemical & Photobiological Sci. 2008. № 11(7). C. 1344.
Hashimoto S., Sunamoto J., Sato K. // Bul. Chem. Soc. Japan. 1967. № 12(40). C. 2860.
Henkelman G., Jónsson H. // J. Chem. Phys. 2000. № 22(113). C. 9978.
Henkelman G., Uberuaga B.P., Jónsson H. // J. Chem. Phys. 2000. № 22(113). C. 9901.
Klamt A., Schüürmann G. // J. Chem. Soc., Perkin Trans. 2. 1993. № 5. C. 799.
Litwinienko G., Ingold K.U. // Accounts of Chem. Res. 2007. № 3(40). C. 222.
Loera D. de, Leyva E., Moctezuma E. // Current Organic Chemistry. 2018. № 15(22). C. 1475.
Lopata K., Govind N. // J. Chem. Theory and Computation. 2011. № 5(7). C. 1344.
Marenich A.V., Cramer C.J., Truhlar D.G. // J. Phys. Chem. B. 2009. № 18(113). C. 6378.
Mikula J.J., Anderson R.W., Harris L.E. // Advances in Molec. Relax. Processes. 1973. № 1–3(5). C. 193.
Ovsyannikov D.V., Zelentsov S.V. // High Energy Chemistry. 2019. № 2(53). C. 103.
Provorse M.R., Isborn C.M. // Intern. J. Quant. Chem. 2016. № 10(116). C. 739.
Rusakowicz B., Testa A.C. // Spectrochim. Acta. Part A: Molec. Spectroscopy. 1971. № 6(27). C. 787.
Trotter W., Testa A.C. // J. Physi. Chem. 1970. № 4(74). C. 845.
Valiev M. et al. Comp. Phys. Communications. 2010. № 9(181). C. 1477.
Winget P. et al. Minnesota Solvent Descriptor Database. Ed. Winget P., Dolney D.M., Giesen D.J., Cramer C.J., Truhlar D.G., Univ. of Minnesota: Minneapolis, 2010.
Wubbels G.G., Jordan J.W., Mills N.S. // J. Am. Chem. Soc. 1973. № 4(95). C. 1281.
York D.M., Karplus M. // J. Phys. Chem. A. 1999. № 50(103). C. 11060.
Zelentsov S.V., Simdyanov I.V. // Rus. Chem. Bul. 2006. № 2(55). C. 207.
Zelentsov S.V., Simdyanov I.V., Kuznetsov M.V. // High Energy Chemistry. 2005. № 5(39). C. 309.
Zugazagoitia J.S., Almora-Díaz C.X., Peon J. // J. Phys. Chem. A. 2008. № 3(112). C. 358.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий