

Химия высоких энергий, 2021, T. 55, № 1, стр. 88-95
Полимеризация акриловой кислоты и метилакрилата в присутствии перекиси бензоила и динитрилаазоизомасляной кислоты в интервале температур, близких к комнатной
А. И. Большаков a, Д. А. Гордон a, *, Н. С. Емельянова a, b, Д. П. Кирюхин a, **
a Институт проблем химической физики Российской академии наук
142432 Черноголовка, Московской обл., просп. Академика Семенова, 1, Россия
b Московский Государственный Университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
* E-mail: diliarag@gmail.com
** E-mail: kir@icp.ac.ru
Поступила в редакцию 18.07.2020
После доработки 19.08.2020
Принята к публикации 02.09.2020
Аннотация
Исследованы кинетика и механизм полимеризации акриловой кислоты и метилакрилата, инициированной перекисью бензоила и динитриломазоизомасляной кислоты в интервале температур 20–40°С. При вакуумировании системы мономер + инициатор процесс полимеризации протекает без нагрева и других методов энергетического воздействия на систему. Выход полимера зависит от концентрации инициатора, температуры и времени термостатирования образцов. Методом ЭПР показано, что мономеры влияют на скорость распада инициатора на радикалы. Квантово-химическими расчетами определены структуры промежуточных комплексов, образующихся в системе акриловая кислота + динитрилазоизомасляной кислоты, посчитаны энергии активации протекающих процессов.
ВВЕДЕНИЕ
Особенность инициаторов распадаться на радикалы при 60–80°С давно известна и широко применяется для инициирования радикальных процессов, в том числе и полимеризации [1–4]. Однако в работе [5] было обнаружено эффективное образование полимера без нагрева или какого-либо иного энергетического воздействия на систему мономер + инициатор. Выход полимера после термостатирования при 23°С в течение 24 ч для большинства исследованных систем достигал 85–90%. Методом ЭПР было показано, что процесс полимеризации радикальный и что мономеры влияют на скорость распада инициатора на радикалы. Исследование оптических спектров (UV, VIS, IR спектроскопия) и квантовохимические расчеты для системы акриловая кислота (АК) + + перекись бензоила (ПБ) показали, что мономер способствует распаду инициатора путем образования между молекулами мономера и инициатора комплексов. В этих комплексах связи, по которым происходит распад инициатора на радикалы, ослабляются. При удалении ингибиторов радикалов, в том числе растворенного в образцах кислорода воздуха, протекает эффективная радикальная полимеризация при комнатной температуре, инициированная радикалами распада инициатора. Было обнаружено, что эффективность реакции резко падает для таких мономеров, как стирол (30%), нонилакрилат (20%), акрилонитрил (50%). В данной работе и в работе [5] и было обнаруженo образованиe полимера (~90%) при введении акриламида в глицерин, содержащий перекисные соединения, образовавшиеся при его хранении, которые играли роль инициатора полимеризации [6, 7]. Однако кинетические особенности такой полимеризации были мало изучены.
Целью настоящей работы явилось исследование кинетики полимеризации АК и МА в присутствии ПБ и динитрилаазоизомасляной кислоты (ДАК) в режиме термостатирования в интервале температур 20–40°С и концентрации инициаторов 0.001–0.3 мол. %.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мономеры акриловую кислоту (СН2=СН–СООН, Тпл = 13°С) и метилакрилат (СН2=СН–СООСН3, Тпл = –75°С) очищали вакуумной перегонкой, отбирая фракции по температуре кипения чистых веществ. ПБ и ДАК трижды перекристаллизовывали из их растворов в метаноле. Растворы ПБ и ДАК в мономерах готовили при комнатной температуре. Полученные растворы вакуумировали (p = 0.3 Па), далее ампулу запаивали и помещали в термостат. После выдерживания образца при определенной температуре ампулу вскрывали, на вакуумной установке удаляли непрореагировавший мономер при 10°С (во избежание дополнительного образования полимера) и гравиметрически определяли выход полимера. ИК спектры регистрировали на ИК-Фурье спектрометре BrukerALPHA, не требующем приготовления образцов в специальных кюветах, при 300 К, в диапазоне 400–4000 см–1. Спектры ЭПР регистрировали на ЭПР спектрометре БрукерElexsys II E 500. Растворы помещали в супрасиловую ампулу с внутренним диаметром 3 мм, длина образца ~25 мм. Число парамагнитных центров в образцах и g-фактор определяли с помощью пакета программ Xepr. Для проверки правильности этих процедур использовали образец CuSO4 ⋅ 5H2O с известной навеской и образец ДФПГ с g-фактором 2.0036. Процедура определения числа парамагнитных центров, используемая в пакете программ Xepr, включает двойное интегрирование спектра.
Квантовохимические расчеты проводились по программе Gaussian 09 версия D [8] с помощью локального функционала B3LYP и базисного набора 6-311++G**. Расчет частот в оптимизированных геометриях исходных и конечных продуктов показал отсутствие мнимых частот, поэтому все оптимизированные геометрии соответствуют минимумам энергии. Для всех полученных переходных состояний расчет частот показал наличие одной мнимой частоты. Для установления истинности переходных состояний были проведены IRC расчеты. Для анализа волновой функции методами QTAIM использовался программный пакет AIMAll (Version 15.05.18) [9]. Расчеты волновых функций и топологических характеристик распределения электронной плотности выполнены в тех же приближениях, что и оптимизация геометрии, B3LYP/6-311++G**. Для вычисления энергии слабых взаимодействий из топологического анализа критических точек связи была использована формула Эспинозы–Лекомпте [10]
(1)
${{E}_{{{\text{A - B}}}}} \approx {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{{\nu }_{e}}(r),$Теоретические ИК спектры моделировались также из расчета частот с помощью программы ChemCraft [11]. Масштабирующий множитель k для метода B3LYP/6-311++G** вычислялся из сравнения теоретического и экспериментального спектров [5] акриловой кислоты и составил 0.944.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 1 представлена зависимость выхода полимера от времени термостатирования АК при 20°С в присутствии 0.01 и 0.015 мол. % ПБ. Выход полимера на начальной стадии медленно возрастает со скоростью 0.01 и 0.075%·мин–1 при содержании ПБ 0.01 и 0.015 мол. % соответственно. После накопления более 10% полимера скорость полимеризации возрастает вплоть до полного превращения мономера в полимер. Аналогичная зависимость наблюдалась для растворов ПБ в МА при 20°С. С увеличением концентрации ПБ кривые зависимости выхода полимера от длительности термостатирования сохраняют свой вид, смещаясь в область меньшего времени термостатирования (рис. 2).
Рис. 1.
Зависимость выхода (%) от времени (мин) термостатирования при 20°С раствора АК + ПБ: 1 – 0.01 мол. % ПБ: 2 – 0.015 мол. % ПБ.
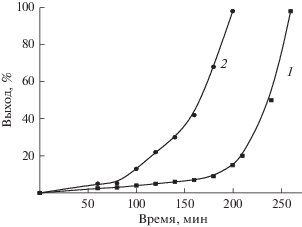
Рис. 2.
Зависимость выхода полимера (%) от времени (мин) термостатирования при 20°С раствора АК + + ПБ: 0.001 мол. % ПБ – 1; 0.01 мол. % ПБ – 2; 0.015 мол. % ПБ – 3; 0.02 мол. % ПБ – 4; 0.03 мол. % ПБ – 5 и раствора АК + 0.015 мол. % ДАК – 6.

При концентрации ПБ 0.03 мол. % реакция переходит сразу же в режим быстрого роста полимерных цепей (~5% мин–1). Схожие результаты были получены и для раствора ДАК в АК. Однако для получения предельного выхода полимера при той же концентрации, как в случае ПБ, требуется более длительное время термостатирования (рис. 2, кривые 3, 6). Это связано, по-видимому, с различной скоростью распада ПБ и ДАК при одинаковой температуре. Следовательно, термостабильность инициатора в его растворе в мономере оказывает заметное влияние на инициирование полимеризации.
При концентрации ПБ ~0.001 мол. % предельный выход полимера ~10% достигается в течение 40 мин и остается постоянным в течение 24 ч термостатирования. Наиболее вероятно, что запределивание выхода полимера при столь малом расходовании мономера связано с полным распадом инициатора за 40 мин. Эти данные позволяют оценить скорость распада инициатора (~1015 молекул см–3 с–1) и скорость роста полимерных цепей по начальному участку кривой накопления полимера (~1017 молекул см–3 с–1). Из полученных результатов следует, что длина полимерной цепи составляет ~102, следовательно, молекулярная масса полимера на начальной стадии роста цепи ~104. Определить молекулярную массу другими методами не представляется возможным, поскольку уже на ранней стадии полимеризации в системе образуется нерастворимый, сшитый полимер. При более высокой концентрации ПБ (более 0.01 мол. %), в условиях быстрого роста полимерных цепей, выход полимера составляет ~95–98%.
Ускорение процесса и столь высокий выход полимера связаны, по-видимому, со стабилизацией активных центров роста цепи по ходу полимеризации из-за сшивания полимерных молекул и формирования сетчатой структуры. Радикальный характер полимеризации АК и МА в присутствии ПБ и ДАК доказан методом ЭПР. Полимеры, полученные на начальной стадии и после завершения процесса полимеризации, нерастворимы и сильно набухают в растворителях. Остановка процесса полимеризации происходит практически при полном расходовании мономера (95–98%). При этом не происходит полной гибели активных радикалов роста цепи (Rp) или полного расходования инициатора (In) в реакции образования полимера, поскольку дополнительное введение мономера в систему после завершения полимеризации приводит к возобновлению реакции (1), что, возможно, связано с “омега-полимеризацией”, наблюдавшейся ранее [12]
(2)
${{{\text{I}}}_{{\text{n}}}} + {\text{ M}} \to 2{\text{R}}_{0}^{\centerdot } + {\text{M}} \to {\text{R}}_{{\text{р}}}^{\centerdot } + {\text{M}} \to {\text{R}}_{{\text{п}}}^{\centerdot },$Данные результаты позволяют предположить, что полученные полимеры, содержащие остаточный инициатор и радикалы полимерных цепей (${\text{R}}_{{\text{п}}}^{\centerdot }$), могут быть использованы в качестве инициаторов для синтеза сшитых привитых блок-сополимеров при добавлении в систему другого мономера (Мх). Схема прививки другого мономера может быть представлена следующим образом:
(3)
${\text{R}}_{{\text{п}}}^{\centerdot } + {{{\text{М}}}_{{\text{x}}}} \to {{{\text{R}}}_{{{\text{px}}}}} + {{{\text{M}}}_{{\text{x}}}} \to {\text{R}}_{{{\text{пx}}}}^{\centerdot },$(4)
${{{\text{I}}}_{{\text{n}}}} + {{{\text{М}}}_{{\text{x}}}} \to 2{\text{R}}_{{{\text{ox}}}}^{\centerdot } + {{{\text{M}}}_{{\text{x}}}} \to {\text{R}}_{{{\text{px}}}}^{\centerdot } + {{{\text{M}}}_{{\text{x}}}} \to {\text{R}}_{{{\text{пx}}}}^{\centerdot }.$Передача цепи и рекомбинация (${\text{R}}_{{{\text{пx}}}}^{\centerdot }$ + ${\text{R}}_{{{\text{пx}}}}^{\centerdot }$) привитых полимерных радикалов, по-видимому, приводит к образованию сетчатой структуры блок-сополимера [1]. Добавленный мономер практически полностью расходуется в прививочной “омега-полимеризации”. Основную роль в инициировании “омега-полимеризации” следует отводить радикалам ${\text{R}}_{{\text{п}}}^{\centerdot }$, стабилизированным сетчатой структурой полимера, поскольку за время реакции инициатор практически полностью расходуется в образовании первичного полимера.
Ускорение полимеризации АК, связанное со стабилизацией макрорадикалов роста цепи, происходит при накоплении в системе ~20% полимера (рис. 3, кривая 1). В случае МА образующийся полимер растворим в собственном мономере, и при полимеризации получается раствор, вязкость которого повышается по ходу процесса. В таком растворе макрорадикалы более подвижны, чем в твердой матрице полимера АК и поэтому ускорение полимеризации, связанное со стабилизацией из-за гель-эффекта растущего макрорадикала ${\text{R}}_{{\text{р}}}^{\centerdot }$, наблюдается при большем выходе (~60%) полимера (рис. 3, кривая 2) и для завершения процесса требуется более длительное время термостатирования. На начальной стадии полимеризации, как видно на кривых рис. 3 (кривые 1, 2), скорости полимеризации различаются незначительно. Это свидетельствует о практически одинаковой скорости распада ПБ как в АК, так и МА, а также активности мономеров в реакции присоединения к растущим макрорадикалам полимерной цепи. Длительное термостатирование (~20 ч) образцов МА и АК с ПБ при температурах ниже 20°С не приводило к образованию полимера. Активность МА и АК в данном случае не является препятствием для образования полимера, поскольку полимеризацию этих мономеров наблюдали ранее [13–15] при более низкой температуре (–163°С). Следовательно, распад ПБ происходит при температурах выше 20°С, при которых и наблюдается образование полимера.
Рис. 3.
Зависимость выхода полимера (%) от времени (мин) термостатирования при 20°С раствора АК + + 0.01 мол. %. ПБ – 1 и МА + 0.01 мол. % ПБ – 2.

На рис. 4 представлена зависимость выхода полимера от температуры для растворов ПБ (концентрация 0.01 мол. %) в АК и МА при времени термостатирования 40 мин. Зарождение полимерных цепей как для АК, так МА, связанное с распадом ПБ, происходит при ~20°С. С повышением температуры выход (и скорость полимеризации) возрастают пропорционально повышению температуры и остаются постоянными до полного превращения мономера в полимер. Разница (примерно в 2 раза) в скоростях образования полимера связана, по-видимому, с различием температурных зависимостей констант роста полимерных цепей МА и АК.
Рис. 4.
Изменение выхода полимера (%) от температуры (Т, °С) растворов АК (1) и МА (2) с перекисью бензоила (0.01 мол. %) при постоянном времени термостатирования 40 мин.

Активность мономера, как отмечалось, не является препятствием в реакции роста полимерных цепей. Несмотря на низкую скорость реакции на начальной стадии, связанную с медленным распадом инициатора, в дальнейшем происходит изменение реакционной среды и ускорение полимеризации. Как уже отмечалось, это явление наблюдали ранее [12] и это явление получило название “омега-полимеризация.”
Период полураспада ПБ при 20°С составляет ~1000 ч [12] что, в принципе, не должно приводить к столь быстрому началу процесса и столь быстрому накоплению полимера (~98%), например, как в случае системы АК+ПБ. Реакция проходит взрывным образом с высоким выходом во время приготовления (вакуумирования) образца. Это означает, что есть процесс, ускоряющий распад инициатора на радикалы. Методом ЭПР мы показали [5], что ускорение процесса распада связано с взаимодействием инициатора с мономером. В раствор ПБ в хлористом метилене ХМ вводили нитроксильный радикал в качестве парамагнитного зонда ПМЗ. За длительное время хранения этой системы изменения концентрации нитроксильных радикалов не наблюдалось. При добавлении мономера в эту систему концентрация нитроксильных радикалов быстро падает до полного исчезновения за счет рекомбинации с радикалами распада ПБ, при этом полимер не образуется. Последнее означает, что реакция рекомбинации радикалов распада с нитроксильным радикалом идет быстрее, чем реакция инициирования полимеризации, а именно реакции присоединения мономера к радикалу распада. Этот эксперимент говорит о том, что мономер способствует распаду ПБ на радикалы. Об этом же говорит эксперимент, из которого следует, что не все мономеры одинаково эффективно полимеризуются в этих условиях. В работе [5] нами обнаружено, что при полимеризации стирола выход полимера составил 30%, нонил-акрилата 20%, акрилонитрила 53%. Анализ литературных данных показал, что наиболее естественно связать это влияние с образованием комплексов между мономером и инициатором, в которых ослабляются связи, по которым осуществляется распад инициатора на радикалы. Авторы [16, 17] методами электронной, ИК, ЯМР спектроскопии и квантовохимических расчетов показали образование комплексов между инициатором ПБ и мономерами акрилонитрилом и малеиновым ангидридом. Установлена их меньшая термостабильность и возможность использования в реакциях синтеза различных соединений. В химии свободных радикалов существует направление исследования радикалообразующих процессов, не требующих для получения радикалов внешнего энергетического воздействия [18, 19]. Разрыв связей происходит за счет перераспределения внутренней энергии системы при образовании комплексов.
В работе [5] квантово-химическими расчетами мы показали, что в растворе ПБ в АК образуются комплексы существенно легче распадающиеся на радикалы, чем ПБ. В данной работе мы провели квантовохимические расчеты для системы ДАК + АК, в которой образование таких комплексов также возможно. Это будет происходить за счет атомов водорода мономера и инициатора и за счет С=О группы мономера или С≡N-группы ДАК. В табл. 1 представлены молекулярные графы оптимизированных геометрий возможных молекулярных комплексов 1, 2, 3, 4, 5, энергии их образования из молекул мономера и инициатора и энергии межмолекулярных связей, вычисленные из метода QTAIM по формуле Эспинозы–Лекомпте.
Таблица 1.
Энергии образования комплексов из молекул мономера и инициатора и энергии межмолекулярных связей
| Молекулярные графы образующихся комплексов | Энергия образования комплекса (B3LYP/6-311++G**) (ккал/моль) |
Энергия межмоле-кулярных связей из метода QTAIM (ккал/моль) | |
|---|---|---|---|
| N…H | O…H | ||
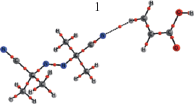 |
1.40 | 1.44 | |
 |
5.97 | 6.43 | 1.29 |
 |
2.55 | 1.22 | 1.95 |
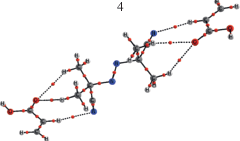 |
6.01 | 1.35 | 1.16 1.48 |
 |
11.84 | 6.43 | 1.19 |
Как видно из табл. 1, энергия всех этих связей лежит в пределах от 1 до 2 ккал/моль. Исключение составляет необычно прочная, 6.43 ккал/моль межмолекулярная связь, образованная за счет С≡N- группы ДАК и водорода карбоксильной группы. Как видно, в данном случае, как и в системе АК + ПБ, могут образовываться комплексы типа М….А и М…А….М, то есть с одной и с двумя молекулами мономера, причем молекула мономера образует от одной до трех межмолекулярных связей. Такие процессы происходят с выигрышем энергии, и чем прочнее образующиеся связи, тем выгоднее образование комплекса. (табл. 1) Самым энергетически выгодным является комплекс 5, в котором имеются две связи С≡N…H–C. При его образовании выделяется вдвое больше энергии, чем для комплекса 4, в котором мономер образует с инициатором три межмолекулярные связи.
Образование молекулярных комплексов в системе мономер-инициатор должно вызывать изменения в ИК спектрах – смещение характеристических полос. Будут смещаться полосы поглощения, отвечающие за колебания связей, которые состоят из атомов, участвующих в образовании межмолекулярных связей.
В табл. 2 приводятся данные ИК спектроскопии исходного мономера (АК) и системы мономер-инициатор, рассматриваются те полосы, которые смещаются в спектре смеси по отношению их положения в спектре мономера. Отнесение полос находится в хорошем согласии с отнесением, приведенным в литературе [20]. В табл. 2 приведены также расчетные данные этих полос для AK и образующегося в системе комплекса 5, как наиболее вероятного молекулярного комплекса, образующегося в смеси AK + ДАK. Как можно видеть из таблицы, расчетные и экспериментальные данные неплохо согласуются между собой.
Таблица 2.
Экспериментальные и теоретические значения положения полос в ИК-спектрах АК
| № | АК | Система мономер-инициатор | Δν/см–1 эксперимен-тальный |
АК | Комплекс 5 | Δν/см–1 теоретический |
Отнесение полос |
|---|---|---|---|---|---|---|---|
| νэксп | νэксп | νтеор | νтеор | ||||
| 1 | 1046 | 1049 | 3 | 1032 | 1047 | 15 | OС–(ОН) + С–Н |
| 2 | 1187 | 1200 | 13 | 1204 | 1250 | 46 | OС–(ОН) |
| 3 | 1358 | 1385 | 27 | 1354 | 1382 | 28 | OС–(ОН) |
| 4 | 3057 | 3062 | 5 | 3044 | 3047 | 3 | СН2=СНst |
Все самые значительные смещения полос колебаний наблюдаются для молекулы мономера, как в эксперименте, так и в квантовохимических расчетах. Основному смещению подвергаются полосы, отвечающие за деформационные плоскостные колебания С(О)ОН-группы (табл. 2). Наличие этих сдвигов связано напрямую с образованием сильной межмолекулярной связи с участием водорода COOH-группы (рис. 5 ). Таким образом, это служит подтверждением образования в системе мономер-инициатор молекулярного комплекса, имеющего строение теоретически предсказанного комплекса 5. То, что расчетное смещение в некоторых случаях несколько больше, связано с тем, что квантовохимические вычисления проводятся для изолированной молекулы.
Таким образом, полагая, что на первой стадии в смеси мономера и инициатора образуется молекулярный комплекс 5, мы можем предложить следующий энергетический профиль реакции полимеризации, включающий в себя инициирование, зарождение и рост цепи (схема 1 ). На первой стадии происходит координация мономера и инициатора за счет водородных связей с образованием молекулярного комплекса. Эта реакция, как было сказано выше, идет с выделением энергии. Далее происходит распад данного комплекса с выделением молекулы азота, то есть по тому же механизму, что и для ДАК. Однако согласно квантовохимическим расчетам, энергетический барьер данной реакции понижается на 3 ккал/моль при образовании молекулярного комплекса. Энергия активации реакции распада ДАК составляет 25.25 ккал/моль, тогда как для молекулярного комплекса 5 она равна 22.50 ккал/моль. В целом данный процесс эндотермический и в результате образуется радикал, также представляющий молекулярный комплекс. Он трансформируется через переходное состояние TS2 (схема 1 ), и таким образом происходит процесс инициирования цепи. Данный шаг идет с не очень значительным энергетическим барьером и является почти термонейтральным. Рост цепи протекает уже со значительным выделением энергии и энергия активации составляет всего 8.17 ккал/моль.
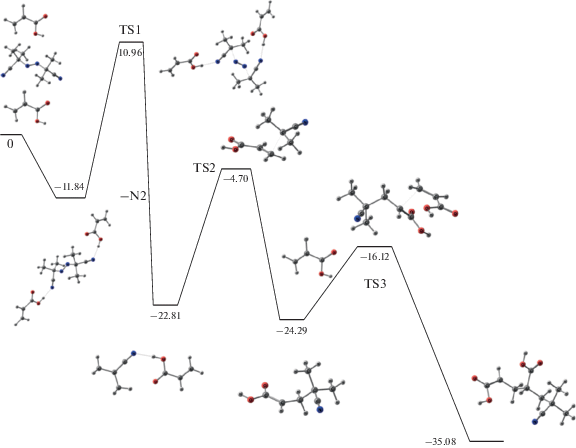
Схема 1 . Энергетический профиль реакции зарождения и роста цепи.
ВЫВОДЫ
Исследована кинетика полимеризации акриловой кислоты и метилакрилата в интервале температур 20–40°С в присутствии вещественных инициаторов – перекиси бензоила и динитрила-азоизомасляной кислоты. Реакция проводилась в вакууме при постоянной температуре в режиме термостатирования. На начальной стадии процесса скорость полимеризации мала. По мере накопления полимера происходит сшивание макромолекул и образование сетчатой структуры, что приводит к стабилизации радикалов роста цепи, т.е. увеличению скорости полимеризации. Таким образом, ускорение процесса полимеризации связано с “омега-полимеризацией”. Выход полимера достигает 95–98%. Стабилизированные макрорадикалы растущих полимерных цепей вместе с остатками вещественного инициатора способны инициировать полимеризацию вновь добавленных мономеров, т.е. могут быть использованы для синтеза привитых сшитых блок сополимеров. С помощью ЭПР, ИК спектроскопии и квантовохимического моделирования показано, что мономер способствует распаду инициатора в системе ДАК + АК, образуя с ним молекулярные комплексы, распадающиеся на радикалы. Предложен энергетический профиль реакции с учетом стадии образования комплексов. Процесс ингибируется кислородом.
Список литературы
Багдасарьян Х.С. Теория радикальной полимеризации. M.: Наука, 1966.
Королев Г.В., Батурина А.А., Березин М.П., Курмаз С.В. // Высокомолек. соед. А. 2004. Т. 46. С. 656.
Froese R., Keaton R., Ewart S., Spencer L., Pearson D., Busch M., Bauer C. // Polymer Engeneering and Science. 2018. V. 59. P. 52.
Генералова А.Н., Ашарчук И.М., Зубов В.П. // Изв. АН, Сер. хим. 2018. Т. 10. С. 1759.
Большаков А.И., Гордон Д.А., Емельянова Н.С., Кузина С.И., Кирюхин Д.П. // Химия высоких энергий. 2019. Т. 53. С. 354.
Bol’shakov A.I., Kiryukhin D.P. // Polym. Sci. Ser. A. 2007. V. 49. P. 975.
Bol’shakov A.I., Kuzina S.I., Kiryukhin D.P. // Polym. Sci. Ser. A. 2008. V. 50. P. 1045.
Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P, Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., J. Peralta E. Jr., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K.N., Staroverov V.N., Keith T., Kobayashi R., NormandJ., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., GompertsR., StratmannR. E., YazyevO., AustinA. J., CammiR., PomelliC., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. // Gaussian, Inc. Wallingford CT. 2013.
AIMAll (Version 15.05.18), Todd A. Keith // TK Gristmill Software, Overland Park KS, USA, 2015 (aim.tkgristmill.com).
Espinosa E., Molins E., Lecomte C. // Chem. Phys. Lett. 1998. V. 285. P. 170.
https://www.chemc raftp rog.com
Уоллинг Ч. Свободные радикалы в растворе. М.: Издательство иностранной литературы, 1960.
Barkalov I., Bolshakov A., Mikhaylov A. // European Polymer J. 1981. V. 17. P. 773.
Гордон Д.А., Эстрина Г.А., Большаков А.И., Михайлов А.И. // Химия высоких энергий. 2015. Т. 49. С. 138.
Гордон Д.А., Кичигина Г.А., Эстрина Г.А. // Химия высоких энергий. 2018. Т. 52. С. 71.
Зайцев С.Ю., Зайцева В.В. Многофункциональные мономеры. Синтез и полимеризация. Донецк: Норд Компьютер. 2003. С. 296.
Зайцев В.В., Тюрина Т.Г. Химия. Раздел 1. 2008. С. 70.
Кузина С.И., Демидов С.В., Денисов Н.Н., Михайлов А.И. // Изв. АН, Cер. хим. 1999. Т. 2. С. 335.
Gordon D.A., Mikhaylov A.I. // e-Polymer. 2015. V. 6. P. 385.
Преч Э., Бюльманн Ф., Аффольтер К. Определение строения органических соединений. М.: Мир, 2006.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий