

Химия высоких энергий, 2023, T. 57, № 1, стр. 14-19
Фотокаталитическое восстановление диоксида углерода в водных суспензиях окисно-титанового полупроводника
Т. С. Джабиев a, *, Л. В. Авдеева a, Т. А. Савиных a, З. М. Джабиева a
a Федеральное государственное бюджетное учреждение Институт проблем химической физики науки
Российской академии наук
142432 Московская обл., Черноголовка, просп. акад. Семенова, 1, Россия
* E-mail: dzhabiev@icp.ac.ru
Поступила в редакцию 30.06.2022
После доработки 19.07.2022
Принята к публикации 19.07.2022
- EDN: DCMNQC
- DOI: 10.31857/S0023119323010047
Аннотация
Изучены реакции фотокаталитического восстановления CO2 в водных суспензиях окисно-титанового полупроводника TiO2 с фотоосажденными сокатализаторами Pt и Cu. Установлено, что состав и количество продуктов восстановления CO2 существенно зависит от природы сокатализатора, нанесенного на TiO2. Предложен механизм образования продуктов восстановления CO2.
ВВЕДЕНИЕ
По мере истощения запасов горючих ископаемых (уголь, нефть, природный газ) все более актуальной становится проблема поиска альтернативных источников энергии, а также сырья для химической промышленности. При сжигании исходного топлива образуется огромное количество диоксида углерода (CO2) и его концентрация в атмосфере Земли постепенно увеличивается. Это может привести к парниковому эффекту, и в конечном итоге, к глобальному потеплению всей планеты. Все это делает весьма актуальной задачу широкомасштабного превращения CO2 в ценные химические соединения. В течение ряда последних десятилетий были сделаны попытки использовать CO2 в качестве исходного вещества в промышленности химического синтеза. И хотя несколько процессов такого рода известны, например, синтез карбамида, соды, салициловой кислоты и т.д., возможности химического использования CO2 до сих пор остаются довольно ограниченными [1]. Усилия же многочисленных исследовательских групп во всем мире направлены на поиск новых реакций с участием CO2. В настоящее время изучаются методы получения органических соединений из CO2 как в фотокаталитических системах, электрохимических, фотоэлектрохимических, в том числе и в присутствии полупроводниковых материалов [2–27 ]. Отметим, что основным недостатком электрохимических и фотоэлектрохимических процессов восстановления CO2 является высокая стоимость потребляемой при этом энергии, которая со временем только возрастет, если в будущем не будут найдены новые источники энергии.
В данной работе приведены результаты исследования фотокаталитического восстановления CO2 в водных суспензиях окисно-титанового полупроводника при осаждении на его поверхности металлических платины (Pt/TiO2) и меди (Cu/TiO2).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные соединения: бидистиллированная вода, SrCl2 · 6H2O “ч”, TiCl4 “ч”, (NH4)2 · SO4 “осч”, H2SO4 “осч”, H2PtCl6 (Merck), CuSO4 “осч”.
Приготовление TiO2 (анатаз)
TiO2 получен по методике [28]. К 100 мл раствора сульфата титана, содержащего 0.9 моль/л Ti(IV) и 1.43 моль/л H2SO4, добавляли 13.7 мл концентрированной H2SO4. После прибавления к этому раствору избытка сульфата аммония (21.7 г), раствор оставляли на 24 ч (сутки) для кристаллизации. Полученный после фильтрования осадок отмывали водой (3 раза) и сушили на воздухе. Затем прокаливали при 750°С 3 ч. Полученный образец имел белый цвет. Нанесение Pt и Cu на поверхность TiO2 проводили по методике [29] фотохимическим восстановлением водного раствора H2PtCl6 и CuSO4 под действием УФ света. На поверхности ПП осаждались высокодисперсные металлические Pt и Cu. В ходе реакции выделялся O2, поскольку донором электронов для восстановления ионов металлов служила H2O. Количество O2 соответствует степени восстановления Me+n.
Фотореакцию проводили в кварцевом реакторе объемом 15 мл с плоским окном диаметром 4 см при облучении ртутной лампой сверхвысокого давления ДРШ-1000. Продукты фотореакции определяли хроматографически с помощью насоса Теплера на хроматографе ЛХМ-8Д, откалиброванных по определяемому веществу. Квантовый выход H2, CO, CH4 определяли по формуле, где W0 – скорость выделения продуктов реакции, I0 – интенсивность светового потока, при λ = 365 нм. Перед использованием CO2 очищали от примесей многократным перемораживанием в вакууме или пропускали через колонку с активированным никель-хромовым катализатором. После очистки во втором случае примесь O2 в реагенте не превышала 0.01 об. %.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Термодинамика процесса восстановления
Известно, что при фотокаталитическом восстановлении CO2, донором электронов является H2O. Одноэлектронное восстановление CO2 водой, т.е. перенос электрона с H2O на CO2 с образование катион-радикала H2O$_{{}}^{{ + \,\,\centerdot }}$ и анион-радикала ${\text{CO}}_{2}^{\centerdot }$ возможно, если уровень зоны проводимости ПП лежит выше одноэлектронного потенциала восстановления CO2 (т.е. выше 1.9 В), а уровень валентной зоны лежит ниже уровня редокс-пары H2O/OH˙ (рис. 1).
Рис. 1.
Положение границ энергетических зон полупроводников и уровней некоторых редокс-пар при Ph 7, по отношению к нормальному водородному потенциалу.
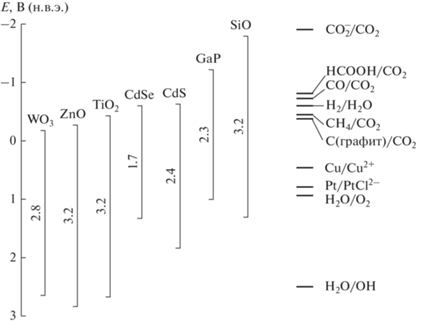
В соответствии со схемой положения границ энергетических зон ПП первое из этих условий не удовлетворяется ни для одной из рассматриваемых ПП, т.е. все уровни зоны проводимости лежат ниже уровня редокс-пары ${\text{CO}}_{2}^{{ - \,\centerdot }}$/CO2. Второму условию удовлетворяют только окисные ПП WO3, ZnO, TiO2. Одноэлектронное восстановление CO2 до ${\text{CO}}_{2}^{{ - \,\,\centerdot }}$ на рассмотренных ПП-материалах невозможно. Однако многоэлектронное восстановление CO2 можно осуществить с образованием таких продуктов, как HCOOH, CO (двухэлектронное), CH2O, С (четырехэлектронное) или CH4 (восьмиэлектронное восстановление). Реакции (1)–(5) с соответствующими редокс-потенциалами относительно нормального водородного редокс-потенциала при pH 7 приведены ниже [30]:
(1)
${\text{C}}{{{\text{O}}}_{2}} + {\text{2}}{{{\text{e}}}^{ - }} + 2{{{\text{H}}}^{ + }} = {\text{ HCOOH}},\,\,\,\,{{E}_{0}} = - 0.61\,\,{\text{В,}}$(2)
${\text{C}}{{{\text{O}}}_{2}} + {\text{2}}{{{\text{e}}}^{ - }} + 2{{{\text{H}}}^{ + }} = {\text{CO + }}{{{\text{H}}}_{{\text{2}}}}{\text{O,}}\,\,\,\,{{E}_{0}} = - 0.53\,\,{\text{В,}}$(3)
$\begin{gathered} {\text{C}}{{{\text{O}}}_{2}} + 4{{{\text{e}}}^{ - }} + 4{{{\text{H}}}^{ + }} = {\text{HCOH + }}{{{\text{H}}}_{{\text{2}}}}{\text{O,}} \\ {{E}_{0}} = - 0.48\,\,{\text{В,}} \\ \end{gathered} $(4)
$\begin{gathered} {\text{C}}{{{\text{O}}}_{{\text{2}}}} + 6{{{\text{e}}}^{ - }} + 6{{{\text{H}}}^{ + }} = {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}, \\ {{E}_{0}} = - 0.38\,\,{\text{В,}} \\ \end{gathered} $(5)
${\text{C}}{{{\text{O}}}_{2}} + 8{{{\text{e}}}^{ - }} + 8{{{\text{H}}}^{ + }} = {\text{C}}{{{\text{H}}}_{4}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}},\,\,\,\,{{E}_{0}} = - 0.24\,\,{\text{В}}{\text{.}}$Известно, что при восстановлении CO2 на металлических электродах из платины образуются CO, HCOOH и углеводороды, а также CH4. Нами исследована фотохимическая реакция восстановления CO2 в водной суспензии катализатора Pt/TiO2, (100 мг TiO2 и 0.5 мас. % Pt в 15 мл H2O), приготовленного фотоосаждением платины на анатаз. На рис. 2 приведены кинетические кривые образования CH4 и H2 при t 30–35°C и pH 6 в водной суспензии.
Рис. 2.
Кинетика выделения CH4 и H2 при фотокаталитическом восстановлении CO2. Условия: 0.1 г TiO2/Pt (0.5 мас. %),CO2 – 1 атм., 303 K, Ph = 6, 20 мл H2O.
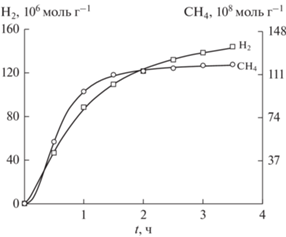
Видно, что процесс образования CH4 через 2–3 ч полностью прекращается, а скорость генерирования H2 существенно снижается. Такое поведение системы полностью согласуется с наблюдавшейся ранее [31] и объясняется отравлением поверхности металлического катализатора монослоем углерода, который осаждался на поверхности металла. Таким образом, была осуществлена эндоэргическая реакция
(6)
${\text{C}}{{{\text{O}}}_{2}} + 2{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{C}}{{{\text{H}}}_{4}} + 2{{{\text{O}}}_{2}},$со стандартной энергией Гиббса ΔG = 1.037 эВ с.
В случае нанесения на поверхность TiO2 металлической меди в качестве катализатора, помимо H2, CH4, C образуется CO. На рис. 3 приведена схема фотокаталитического восстановления СО2 водой на Cu/TiO2. Кинетические кривые образования CO в зависимости от количества взятого катализатора Cu/TiO2 представлены на рис. 4. Количество катализатора Cu/TiO2 меняли от 0.08 до 0.14 г.
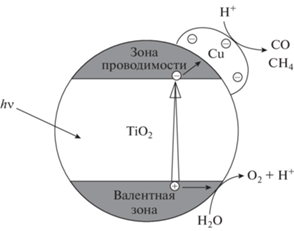
Рис. 4.
Зависимость скорости выделения CO при фотокаталитическом восстановлении CO2 водой от времени. Условия: TiO2/Cu (0.5 мас. %), CO2 – 1 атм, 1 – 0.08 , 2 – 0.1, 3 – 0.14 г, 303К, Ph = 6, 20 мл H2O.
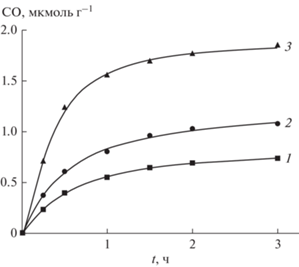
Оксид углерода образуется без индукционного периода. При увеличении количества взятого ПП Cu/TiO2 от 0.08 г до 0.14 г начальная скорость генерирования CO увеличивается в 7 раз, а выход CO за 3.5 ч до 1.85 мкмоль · г–1. Квантовый выход CO Ф = 0.04.
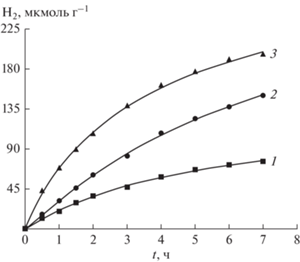
Аналогичным образом ведет себя и кинетика образования H2 в тех же условиях фотохимической реакции восстановления, представленная на рис. 5, причем начальные скорости образования H2 меняются симбатно (как и в случае CO). Они равны соответственно 20, 33 и 75 мкмоль/ч, что согласуется с отношением скоростей выделения CO – продукта двухэлектронного восстановления CO2 (рис. 4). Оцененный квантовый выход H2, Ф = 0.22.
Рис. 5.
Кинетика выделения H2 при фотокаталитическом восстановлении CO2 водой от времени. Условия (см. рис. 4). Кривая 3 – образец после потери активности прогревался в токе воздуха при 773 К в течение двух часов.
В отличие от кинетических кривых образования H2 и CO, кинетика накопления CH4, приведенная на рис. 6, имеет S-образный вид, что указывает на стадийный механизм формирования CH4, причем окись углерода CO не является предшественником образования CH4, поскольку максимальная концентрация CН4 достигается не через 2.5 ч, как это имеет место в случае СО (см. рис. 4).
Рис. 6.
Кинетика выделения CH4 при восстановлении CO2 водой в разных условиях приготовления катализатора. Условия: количество взятого TiO2/Cu: 1 – 0.1 , 2 – 0.14, 3 – 0.08 г; СО21 – 0.75, 2, 3 – 1 атм; кривая 1 – образец дополнительно насыщался CO2 (указано стрелкой), кривая 3 – образец прогревался после потери активности в течение двух часов в токе воздуха при 773 K.
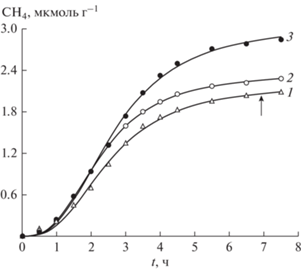
Снижение скорости образования CH4 после 4–5 ч фотолиза не связано с израсходованием предварительно адсорбированного на поверхность ПП CO2, поскольку добавление в реагирующую систему в ходе реакции дополнительных количеств CO2 (указано стрелкой на кривой 1 рис. 6) практически не меняет закономерного снижения активности катализатора.
Разумеется, скорость генерирования продуктов восстановления должна зависеть от количества адсорбированного в начале реакции CO2. Кривая 1 на рис. 6 (начальный участок) получена при проведении фотокаталитической реакции на хранившемся фотокатализаторе на воздухе в течение суток. За это время фотокатализатор (Cu/TiO2) адсорбировал почти предельно возможное количество CO2, хотя его концентрация в окружающей среде составляет ~0.03%.
Поскольку на кривой 1 (рис. 6) вначале в системе присутствовало несколько меньше адсорбированного CO2, то максимальная скорость образования CH4 оказалась меньше на 25%. Выход же CH4 после напуска в реактор дополнительного количества CO2 практически равен выходу на кривой 2 (рис. 6), когда количество взятого катализатора (Cu/TiO2) ~ на 18% превосходило соответствующее количество на кривой 1. Возможно, это связано с большим количеством образовавшегося в случае кривой 2 (рис. 4) монооксида углерода CO (кривая 2 на рис. 4) по сравнению с кривой 1. Полная утрата каталитической активности фотокатализаторов Cu/TiO2 (кривые 2 и 3 на рис. 6 после 7 ч фотолиза) также объясняется отравлением катализатора, т.е. покрытием металлической поверхности Cu в фотопроцессе углеродом, по-видимому, в виде графита.
Полное отравление катализатора формирования CH4 не приводит к полной остановке процесса образования H2 после 7 ч фотолиза (рис. 5). Поскольку катализатором обоих процессов является металлическое покрытие (Pt или Cu), то продолжение образования H2 хотя и с меньшей скоростью, когда CH4 уже не образуется, связано с меньшими стерическими затруднениями для малой молекулы H2 покинуть поверхность металла, почти закрытую плотным слоем графита. Однако мы полагаем, что активные центры формирования H2, CO и CH4 при фотовосстановлении СО2 различны. Несмотря на то, что они образованы на одном и том же катализаторе наблюдается большая разница в начальных скоростях образования H2 (см. кривые 2, 3 на рис. 5) при практически совпадающих начальных скоростях образования CH4 в тех же условиях (см. рис. 6). Прямым подтверждением отравления катализатора углеродной пленкой в ходе процесса служит регенерация исходной активности термической обработкой в токе воздуха.
Кривые 3 на рис. 6 и 5 представляют кинетику образования CH4 и H2, которые после потери каталитической активности прогревались в течение двух часов в токе воздуха при 773 К. Видно, что активность в формировании CH4 восстановилась полностью, тогда как для H2 и CO-генерирующей способности недостаточно. Эффективность образования CH4 достигает Ф = 0.012.
ВОЗМОЖНЫЙ МЕХАНИЗМ ОБРАЗОВАНИЯ ПРОДУКТОВ ФОТОКАТАЛИТИЧЕСКОГО ВОССТАНОВЛЕНИЯ CO2 ВОДОЙ
Согласно нашим исследованиям CO2 связывается с поверхностью меди лишь слабыми силами физической адсорбции. Не было замечено никаких эффектов, результатом которых была бы диссоциация адсорбированного CO2, хотя авторы работы [32] не исключают полностью возможности образования промежуточных продуктов диссоциации, что может способствовать избыточному обмену. Механизм восстановления CO2 не может включать прямого переноса ${\text{e}}_{{{\text{з}}{\text{.п}}}}^{ - }$ на адсорбированную молекулу CO2, поэтому надо думать, что первая стадия восстановления состоит в реакции фотогенерированных хемисорбированных атомов водорода на медной поверхности, аналогично тому, как это предполагается для электрохимического процесса [33]. Последующие стадии ведут к адсорбированным на поверхности молекулам CO, которые могут десорбироваться в газовую фазу или подвергаться дальнейшему восстановлению на поверхности Cu до CH4.
Эти стадии можно записать следующим образом:
(7)
${{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{Cu}} + {\text{e}}_{{{\text{з}}{\text{.п}}{\text{.}}}}^{ - } \rightleftarrows {\text{Cu--}}{{{\text{H}}}_{{{\text{адс}}{\text{. + }}}}}{\text{O}}{{{\text{H}}}^{ - }},$(8)
${\text{Cu}} + {\text{C}}{{{\text{O}}}_{2}} \rightleftarrows {\text{Cu}}{{\left( {{\text{C}}{{{\text{O}}}_{{\text{2}}}}} \right)}_{{{\text{физ}}{\text{.адс}}{\text{.}}}}},$(9)
${\text{Cu--}}{{{\text{H}}}_{{{\text{адс}}{\text{.}}}}} + {\text{Cu}}{{\left( {{\text{C}}{{{\text{O}}}_{{\text{2}}}}} \right)}_{{{\text{физ}}{\text{.адс}}{\text{.}}}}} \to {\text{Cu--OCH}}{{{\text{O}}}_{{{\text{адс}}{\text{.}}}}},$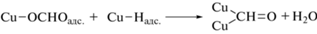
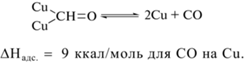
Один из часто наблюдаемых продуктов восстановления –HCOOH может быть получен при акцептировании второго ${\text{e}}_{{{\text{з}}{\text{.п}}}}^{ - }$ промежуточным продуктом реакции (3)
(12)
${\text{Cu--OCH}}{{{\text{O}}}_{{{\text{адс}}{\text{.}}}}} + {\text{e}}_{{{\text{з}}{\text{.п}}}}^{ - } \to {\text{Cu}} + {\text{HCO}}{{{\text{O}}}^{ - }}.$Образование же продуктов, идентифицируемых в фотокаталитическом процессе, можно представить следующим образом:
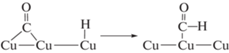
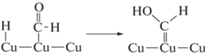
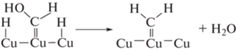
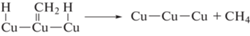
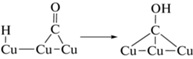
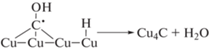
Фиксируемый на начальных стадиях фотопроцесса CO образуется по реакции (11). Последовательное восстановление хемисорбированного CO на поверхности меди приводит либо к образованию CH4 по реакциям (13)–(16), либо на поверхности осаждается углеродная пленка по реакциям (17)–(18), что приводит к отравлению катализатора. Регенерация сводится к взаимодействию поверхностного углерода с кислородом (выжигание), с освобождением активных центров от углерода. Молекулярный водород образуется при взаимодействии двух частиц типа Cu–Hадс. Таким образом, могут быть объяснены все наблюдавшиеся маршруты этого сложного процесса.
ВЫВОДЫ
1. Показано, что металлическая медь может служить катализатором формирования продуктов H2, CO, CH4 фотокаталитического восстановления CO2 в водных суспензиях окисно-титанового полупроводника TiO2.
2. Выход продуктов восстановления CO2 на катализаторе TiO2/Cu выше, чем на катализаторе TiO2/Pt.
3. Предложен механизм образования продуктов восстановления CO2 водой.
Список литературы
Катализ в С1-химии / Под. ред. Кайма В. Л.: Химия, 1987. 296 с.
Zhong W., Sa R., Li L., et all // J. Am. Chem. Soc. 2019. V. 141. P. 7615. https://doi.org/10.1021/jacs.9b02997
White J.L., Baruch M.F., Pander III J.E., et all // Chem. Rev. 2015. V. 115. P. 12888. https://doi.org/10.1021/acs.chemrev.5b00370
Li X., Yu J., Jaroniec M., Chen X. // Chem. Rev. 2019. V. 119. P. 3962. https://doi.org/10.1021/acs.chemrev.8b00400
Takeda H., Cometto C., Ishitani O., Robert M. // ACS Catal. 2017. 7. P. 70. https://doi.org/10.1021/acscatal.6b02181
Francke R., Schille B., Roemelt M. // Chem. Rev. 2018. V. 118. P. 4631. https://doi.org/10.1021/acs.chemrev.7b00459
Rao H., Schmidt L., Bonin J., Robert M. // Nature. 2017. V. 548. P.74. https://doi.org/10.1038/nature23016
Fang Y., Wang X. // Chem. Commun. 2018. V. 54. P. 5674. https:doi.org/https://doi.org/10.1039/C8CC02046A
Maeda K., Kuriki R., Zhang M., et all // J. Mater. Chem., A. 2014. 2. P. 15146. https://doi.org/10.1039/C4TA03128H
Kuhl K.P., Cave E.R., Abram D.N., Jaramillo T.F. // Energy Environ. Sci. 2012. 5. P. 7050. https://doi.org/10.1039/C2EE21234J
Arquer F.P.G.D., Bushuyev O.S., Luna P.D., et all // Adv. Mater. 2018. 30. 1802858. https://doi.org/10.1002/adma.201802858
Gao S., Lin Y., Jiao X., et all // Nature. 2016. V. 529. P. 68. https://doi.org/10.1038/nature16455
Jouny M., Luc W., Jiao F. // Ind. Eng. Chem. Res. 2018. V. 57. P. 2165. https://doi.org/10.1021/acs.iecr.7b03514
Kuilin Lv., Yanchen Fan, Ying Zhu, et all // J. Mater. Chem., A. 2018. V. 6. № 12. P. 5025. https://doi.org/10.1039/C7TA10802H
Lee S., Park G., Lee J. // ACS Catal. 2017. 7. P. 8594. https://doi.org/10.1021/acscatal.7b02822
Xu S., Carter E.A. // J. Am. Chem. Soc. 2018. V. 140. 28. P. 8732. https://doi.org/10.1021/jacs.8b03774
Brown E.S., Peczonczyk S.L., Wang Z., Maldonado S. // J. Phys. Chem., C. 2014. V. 118. 22. P. 11593. https://doi.org/10.1021/jp503147p
Beiler A.M., Khusnutdinova D., Jacob S.I., Moore G.F. // ACS Appl. Mater. Interfaces. 2016. 8. 15. P. 10038. https://doi.org/10.1021/acsami.6b01557
Keith J.A., Carter E.A. // J. Am. Chem. Soc. 2012. V. 134. 18. P. 7580. https://doi.org/10.1021/ja300128e
Lu X., Huang S., Diaz M.B., et all // IEEE Journal of Photovoltaics. 2012. 2. 214. https://doi.org/10.1109/JPHOTOV.2011.2182180
Navalón S., Dhakshinamoorthy A., Álvaro M. // ChemSusChem. 2013. P. 562. https://doi.org/10.1002/cssc.201200670
Huygh S., Bogaerts A., Neyts E.C. // J. Phys. Chem., C. 2016. V. 120. 38. P. 21659. https://doi.org/10.1021/acs.jpcc.6b07459
Yongfei Ji, Yi Luo // J. Am. Chem. Soc. 2016. V. 138. 49. P. 15896. https://doi.org/10.1021/jacs.6b05695
Xie S., Wang Y., Zhang Q., et all // Chem. Commun. 2013. 49. P. 2451. https://doi.org/10.1039/C3CC00107E
White J.L., Baruch M.F., Pander III J.E., et all // Chem. Rev. 2015. V. 115. 23. P. 12888. https://doi.org/10.1021/acs.chemrev.5b00370
Chang X., Wang T., Gong J. // Energy Environ. Sci. 2016. 9. P. 2177. https://doi.org/10.1039/C6EE00383D
Mao J., Li K., Peng T. // Catal. Sci. Technol. 2013. 3. № 10. P. 2481. https://doi.org/10.1039/C3CY00345K
Горощенко Я.Г. Химия титана. Киев: Наукова думка, 1970. 416 с.
Lehn J.-M., Sauvage J.-P., Ziessel R. // Nouv. J. Chim. 1984. V. 4. № 11. P. 623.
Dimitrijevic N.M., Vijayan B.K., Poluektov O.G., et al. // J. Am. Chem. Soc. 2011. V. 133. P. 3964. https://doi.org/10.1021/ja108791u
Hemminger J.C., Carr R., Somorjai G.A. // Chem. Phys. Lett. 1978. V. 57. 1. P. 100. https://doi.org/10.1016/0009-2614(78)80359-5
Haas T., Pritchard J. // J. Chem. Soc., Faraday Trans. 1990. V. 86. № 10. P. 1889. https://doi.org/10.1039/FT9908601889
Cook R.L., MacDuff R.C., Sammells A.F. // Electrochem. Soc. 1988. V. 135. № 6. P. 1320. https://doi.org/10.1149/1.2095972
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий