

Микроэлектроника, 2022, T. 51, № 6, стр. 429-442
Моделирование адсорбции золота на поверхность дефектного графена
М. М. Асадов 1, 2, *, С. О. Маммадова 3, С. С. Гусейнова 3, С. Н. Мустафаева 3, В. Ф. Лукичев 4, **
1 Институт катализа и неорганической химии им. М.Ф. Hагиева Национальной
академии наук Азербайджана
AZ-1143 Баку, пр. Г. Джавида, 113, Азербайджан
2 Научно-исследовательский институт геотехнологических проблем нефти,
газа и химии АГУНП
AZ-1010 Баку, пр. Азадлыг, 20, Азербайджан
3 Институт физики Национальной академии наук Азербайджана
AZ-1143 Баку, пр. Г. Джавида, 131, Азербайджан
4 Физико-технологический институт им. К.А. Валиева Российской
академии наук
117218 Москва, Нахимовский просп., 36, корп. 1, Россия
* E-mail: mirasadov@gmail.com
** E-mail: lukichev@ftian.ru
Поступила в редакцию 31.05.2022
После доработки 09.07.2022
Принята к публикации 09.07.2022
- EDN: ABYBTP
- DOI: 10.31857/S054412692270003X
Аннотация
Представлены результаты теоретического исследования локальных структурных изменений и адсорбционных характеристик поверхности графена (GP) в присутствии комплекса “вакансия + адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$”. На основе теории функционала плотности (DFT) рассчитаны адсорбционные свойства ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на поверхности графеновых суперъячеек, содержащих 50 атомов углерода с вакансиями $({\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle ,{\text{\;G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $). Определена стабильная конфигурация суперъячеек ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с комплексом “вакансия + адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$”. Рассчитано влияние адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на зонную структуру и локальный магнитный момент в ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. Анализ электронной структуры проводился на основе равновесной атомной конфигурации ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $, локальной плотности электронных состояний и с учетом спиновой поляризации. Расчеты проводились с использованием обменно-корреляционного функционала в приближении локальной электронной спиновой плотности (LSDA).
1. ВВЕДЕНИЕ
Графен (${\text{GP}}$) исследуется как перспективный материал, например, для формирования двумерной подложки, в наноэлектронике и спинтронике. Известны экспериментальные [1–3] и теоретические [4–10] данные об исследовании адсорбции различных металлов, в том числе и золота, на поверхности графена.
Адсорбция атомов (адатом) металлов на поверхность графена является эффективным способом поляризации носителей заряда [2]. Изучение магнетизма в графене показало, что формирование локального магнитного момента в ${\text{GP}}$ зависит как от концентрации, так и от геометрии дефектов на поверхности [3].
Ab initio расчеты адсорбции металлов, в частности, золота (${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$) на поверхность графена ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ [4–10] показывают, что вычисленные характеристики (энергия адсорбции [4–8], магнитный момент [6]) зависят от различных факторов: от метода расчета [5, 9], структуры адатома [7], места адсорбции (сверху, мостовое и полое положения) [4, 5], расстояния связи ($d$) между адатомом и поверхностью [5, 8], характера взаимодействия между адатомом и графеном [6–9], электронной структуры подложки GP [9, 10].
В известных работах по адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на поверхность ${\text{GP}}$ в основном изучали бездефектный ${\text{GP}}$ с использованием разных обменно-корреляционных функционалов (Exchange-correlation Functionals – ${{E}_{{{\text{XC}}}}}$). Однако, как известно, поверхностные дефекты, заметно влияют на электронные, магнитные, механические свойства поверхности. С учетом этого актуальным является исследование адсорбционных и электронных свойств поверхности в присутствии комплекса “вакансия + адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$” структуры ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
Цель работы – Ab-initio расчет энергии адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на поверхности графеновых суперъячеек ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с точечными дефектами (${\text{V}}$ – вакансия).
2. МОДЕЛЬ И МЕТОДИКА РАСЧЕТОВ
Расчеты зонной структуры дефектного графена проводили на основе теории функционала плотности (DFT). Исследовали суперъячейки Au-адсорбированного графена ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $, содержащего вакансию. Расчеты проводили с помощью программного пакета Atomistix ToolKit при температуре T = 0 K [11, 12]. Использовали суперъячейки ${\text{GP}}$, содержащие 50 атомов углерода.
Электронная конфигурация свободного атома важна для правильного описания химической связи в системе. Использовались следующие электронные конфигурации для атомов: Au –$\left[ {{\text{Xe}}} \right]4{{f}^{{14}}}~4{{d}^{{10}}}~4{{s}^{1}}$, C – $\left[ {{\text{He}}} \right]2{{s}^{2}}2{{p}^{2}}$. Состояния $\left[ {{\text{Xe}}} \right]$и $\left[ {{\text{He}}} \right]$ относятся к остовным. Основное состояние как атома золота $(4{{f}^{{14}}}~4{{d}^{{10}}}~4{{s}^{1}}$), так и углерода ($2{{s}^{2}}2{{p}^{2}})$ являются состоянием с открытой оболочкой. Для таких состояний учитывали эффекты спиновой поляризации.
Обменно-корреляционный функционал ${{E}_{{{\text{XC}}}}}$ рассчитывали в приближении локальной электронной спиновой плотности (LSDA) в параметризации Пердью–Цунгера [13]. В расчетах использовали постоянную решетки графена, равную ее оптимизированному значению. Для интегрирования в обратном пространстве использовалась схема Монхорста–Пака [14] с сеткой из 5 × 5 × 5 k-точек в зоне Бриллюэна.
Расчет электронной структуры суперъячеек размером 5 × 5 ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ проводили с учетом основных состояний атомов ${\text{Au}}$ и углерода в k-точках выборки суперъячейки графена в зоне Бриллюэна по линиям M-Γ-K-M. Это позволяет проследить за дисперсией точек Дирака на краях зоны Бриллюэна.
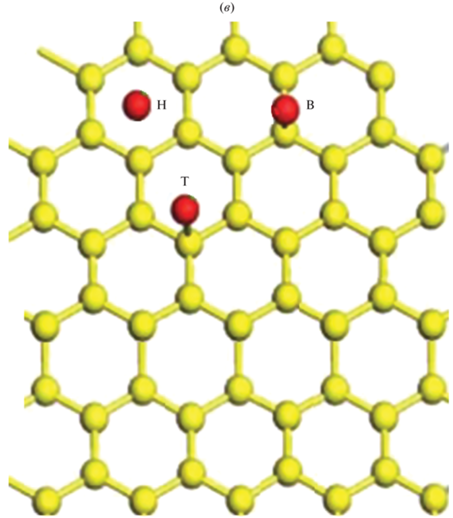
DFT-расчеты проводили для трех разных центров адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}\left( {111} \right)$ на графене [4]: мостиковое положение B-сайт (${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен в мостиковом участке между С–С связи на ${\text{GP}}$), углубления в центре H-сайт (${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен в центре шестичленного кольца С–С связи на ${\text{GP}}$) и расположение адатома сверху T-сайт (${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен в тетраэдрической конфигурация С–С связи на ${\text{GP}}$) (рис. 1).
Рис. 1.
Адсорбционные центры 5 × 5 суперъячейки монослоя графена. B-сайт (мостиковое расположение; расположение в центре связи C–C); T-сайт (тетраэдрический сайт; расположение сверху); H-сайт (гексагональный сайт; расположение в центре шестичленного кольца). а – бездефектный графен, б – графен с одной вакансией, в – графен с одной вакансией и адатомами.
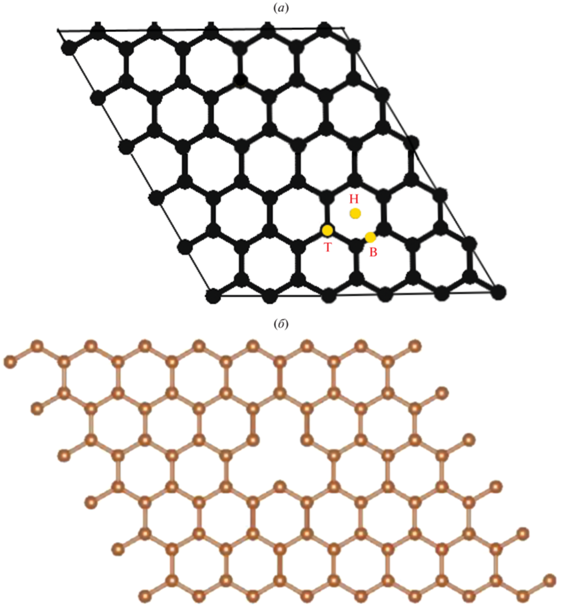
Рис. 1.
Окончание.
Расчеты энергии адсорбции адатомов ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ и магнитных свойств на поверхности графеновых структур проводили в оптимизированных суперъячейках GP. Кинетическая энергия отсечки при обменной корреляции составляла 500 эВ, которая обеспечивала сходимость по полной энергии 10–5 эВ/ячейка. Каждую исследуемую суперъячейку предварительно релаксировали с допусками максимальной силы 0.01 эВ/Å, напряжения 0.01 эВ/Å3 и максимальным смещением 0.001 Å соответственно.
Энергию адсорбции адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ рассчитывали по формуле [12]
(1)
$\begin{gathered} E_{{{\text{ads}}}}^{{{\text{atom}}}} = \frac{1}{{m + n}}~{{E}_{{{\text{total}}}}}\left( {{\text{graphene}} + {\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right) - \\ - \,\,\left[ {mE_{{{\text{iso}}}}^{{\text{C}}}\left( {{\text{graphene}}} \right) + nE_{{{\text{iso}}}}^{{{\text{Au}}}}\left( {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right)} \right], \\ \end{gathered} $Деформацию монослоя графена при адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}\left( {111} \right)~$ рассчитывали по формуле: $\frac{{d_{{{\text{C}} - {\text{C}}}}^{{{\text{free}}}} - d_{{{\text{C}} - {\text{C}}}}^{{{\text{ads}}}}}}{{d_{{{\text{C}} - {\text{C}}}}^{{{\text{free}}}}}}~ \times 100$, где $d_{{{\text{C}} - {\text{C}}}}^{{{\text{free}}}}$ и $d_{{{\text{C}} - {\text{C}}}}^{{{\text{ads}}}}$ – длины связи C–C в свободном и адсорбированном монослоях графена.
Полные и парциальные магнитные моменты оценены по известной методике. Обменные интегралы в гамильтониане учтены в следующем виде: $H = ~ - \sum\nolimits_{i \ne j} {{{J}_{{ij}}}{{{\text{S}}}_{i}}{{{\text{S}}}_{j}}} $, где ${{J}_{{ij}}}$ – параметры магнитного обменного взаимодействия между атомами i и j, ${{{\text{S}}}_{i}}$ – полный спин атома i. Магнитный момент атома i связан со спином ${{{\text{S}}}_{i}}$ следующим соотношением: ${{M}_{i}} = g{{\mu }_{{\text{B}}}}{{{\text{S}}}_{i}}$, где $g$ – фактор Ланде (гиромагнитный множитель), ${{\mu }_{{\text{B}}}}$ – магнетон Бора (элементарный магнитный момент).
3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В графене, как известно, каждый атом углерода связан с тремя другими атомами углерода. В соответствии с sp2-гибридизацией атома углерода, который имеет два 2s- и два 2p-электрона в валентном состоянии, длина связи С–С в графене меняется.
Согласно DFT-расчетам с использованием ${{E}_{{{\text{XC}}}}}$ LDA (аппроксимация локальной плотности) длина связи С–С в чистом графене (1.41 Å), была меньше, чем длина связи С–${\text{Au}}$ в изученных нами структурах ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
3.1. Адсорбция
Энергия связи $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ адатома с поверхностью зависит, в частности, от длины связи ($d$) между адатомом и поверхностью. Очевидно, что если среднее расстояние $d$ большое, то $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ должна быть малой. В случае большого расстояния связи между адатомом и поверхностью проявляется физическая адсорбция. Если расстояние связи короткое, то $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ должна увеличиться за счет химической адсорбции.
Адсорбция Au на GP зависит от времени пребывания $\left( t \right)$ адатома и его пройденного расстояния $\left( \lambda \right)$ до подложки перед десорбцией. Эти величины связаны с энергиями атомов для адсорбции $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ и поверхностной диффузии $E_{{\text{d}}}^{{{\text{atom}}}}$ соотношением
(2)
$\tau = ~\nu _{{{\text{ads}}}}^{{ - 1}}~{\text{exp}}\left( {\frac{{E_{{{\text{ads}}}}^{{{\text{atom}}}}}}{{{{k}_{{\text{B}}}}T}}} \right),$(3)
$\lambda = ~\frac{a}{2}~\sqrt {\frac{{{\text{\;}}{{\nu }_{{\text{d}}}}}}{{{\text{\;}}{{\nu }_{{{\text{ads}}}}}}}} ~{\text{exp}}\left( {\frac{{E_{{{\text{ads}}}}^{{{\text{atom}}}} - E_{{\text{d}}}^{{{\text{atom}}}}}}{{2{{k}_{{\text{B}}}}T}}} \right),$Атомные структуры ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ предварительно релаксировали методом DFT LDA. Первоначально атом ${\text{Au}}$ помещался на расстоянии 2.5 Å от поверхности графена. А монослой графена был “заморожен”. В конце релаксации сумма всех сил, действующих в системе, составляла ≤0.001 эВ/Å. Атомные структуры ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ для трех различных конфигураций (B-сайт, H-сайт и T-сайт) расположения ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ после релаксации сравнивали.
Деформация монослоя графена при адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ незначительная. Установленные нами равновесные параметры решеток, позиции атомов углерода в графене и адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ согласуются с данными [4–7]. Определены длины связей между атомами углерода в графене, дистанции между парами атомов ${\text{Au}}{\kern 1pt} - {\kern 1pt} {\text{C}}$ для систем ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ после релаксации. В системе ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ среднее расстояние между атомами углерода и золота, составило $d_{{{\text{eq}}}}^{{\text{T}}}$ = 2.56 Å для места связывания атомов ${\text{Au}}{\kern 1pt} - {\kern 1pt} {\text{C}}$ сверху (T-сайт). Это согласуется с данными $d_{{{\text{eq}}}}^{{\text{T}}}$ = 2.61 Å [7].
На рис. 2 показана схема оптимизированных нами 5 × 5 суперъячеек ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle .$ В этом случае энергия связи $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ характеризует разность энергий релаксированной структуры ${\text{GP}}$ по отношению к той же структуре ${\text{GP}}$, в которой атом Au находится в вакууме.
Рис. 2.
Геометрическая модель расположения адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на монослое 5 × 5 суперъячейки графена, содержащей 50 атомов углерода: (а) суперъячейка графена ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположенным в мостиковом участке (B-сайт) между С–С связью, (б) суперъячейка дефектного графена ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с одной вакансией, где ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен в мостиковом участке (B-сайт) между С–С связью, (в) суперъячейка дефектного графена ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с одной вакансией, где ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен сверху (T-сайт) над атомом углерода.
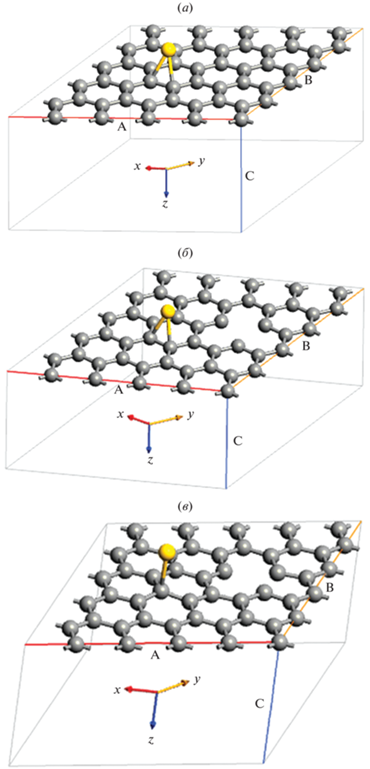
Начальное расположение адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}\left( {111} \right){\text{\;}}$ на поверхности ${\text{GP}}$ после релаксации системы ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ незначительно изменяется. Это связано с тем, что адсорбированный атом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}\left( {111} \right){\text{\;}}$создает область локального сжатия поверхности с вакансией ${\text{G}}{{{\text{P}}}_{{\text{V}}}}$.
Сравнивали энергии адсорбции $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ для ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$, адсорбированного по трем позициям: сверху (T-сайт), мостовой (B-сайт) и полой (H-сайт). Для систем ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ более стабильными оказались конфигурации связывания атомов ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}{\kern 1pt} - {\kern 1pt} {\text{C}}$ адатом сверху (T-сайт).
Расчеты $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ адатомов ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на поверхности графена с использованием функционала LDA показывают чуть завышенные значения $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ по сравнению с LSDA данными$.$ Учет спиновой поляризации в LSDA расчетах позволяет корректировать LDA расчетную величину $E_{{{\text{ads}}}}^{{{\text{atom}}}}$.
Учитывая спиновое расщепление атома золота, корректировали также энергию обменного взаимодействия в ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. В этом случае LSDA-расчетная величина $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ уменьшается, что находится в согласии с расчетами [4, 5].
В табл. 1 приведены результаты наших DFT-LSDA расчетов для 5 × 5 суперъячеек ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ на основе графена в сопоставлении с известными данными.
Таблица 1.
DFT LSDA рассчитанные энергии адсорбции адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ (эВ/атом) на 5 × 5 суперъячейках ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и равновесное среднее расстояние ($d_{{{\text{eq}}}}^{{\text{T}}}$) между атомами ${\text{Au}}{\kern 1pt} - {\kern 1pt} {\text{C}}$. Данные LSDA показаны для места связывания атомов ${\text{Au}}{\kern 1pt} - {\kern 1pt} {\text{C}}$, где адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен сверху (T-сайт) поверхности
| Структура, ${{E}_{{{\text{XC}}}}}$ функционал | $E_{{{\text{ads}}}}^{{\text{T}}}$, ${\text{эВ/атом}}$ | $d_{{{\text{eq}}}}^{{\text{T}}}$, $~{\text{{\AA}}}$ |
|---|---|---|
| ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ | ||
| LDA [4] | –0.77 | >2 |
| LDA [5] | –0.732 | 2.22 |
| GGA [6] | –0.107 | 2.82 |
| GGA [7] | –0.310 | 2.61 |
| PBE [8] | –0.51 | 2.33 |
| LSDA | –0.41 | 2.56 |
| ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ | ||
| LSDA | –3.36 | 2.21 |
Результаты расчета $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ для суперъячейки на основе бездефектного графена ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ аналогичны результатам работ [4, 5]. $E_{{{\text{ads}}}}^{{{\text{atom}}}}$ адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ в мостовой (B-сайт) и полой (H-сайт) положениях вырождены, т.е. ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ может легко диффундировать сверху на поверхность ${\text{GP}}$ в системе ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
Рассчитанное равновесное расстояние $d_{{{\text{eq}}}}^{{\text{T}}}\left( {\text{{\AA}}} \right)$ между адатомом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ и ближайшим атомом C на плоскости графена составляет 2.56 ${\text{{\AA}}}$, что согласуется с данными [4–8]. Вычисленное расстояние $d_{{{\text{eq}}}}^{{\text{T}}}\left( {\text{{\AA}}} \right)$ мало зависит от функционала, используемого в DFT-расчете. Значение $d_{{{\text{eq}}}}^{{\text{T}}}\left( {\text{{\AA}}} \right)$ меньше для системы с моновакансией графена ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $, чем для бездефектного графена ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
Как видно из табл. 1, энергия адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на графене с одиночной вакансией ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ почти на порядок меньше, чем на “чистом” графене ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. Этот эффект стабилизации структуры обусловлен гибридизацией d-орбиталей Au: [Xe] $4{{f}^{{14}}}~4{{d}^{{10}}}~4{{s}^{1}}$ с оборванными связями sp2, присутствующими в соседних атомах углерода ${\text{GP}}$, с одиночной вакансией.
3.2. Энергия образования дефекта
В реальных условиях любой кристалл не является идеальным, а содержит различные дефекты. А свойства, в частности, механические, электрофизические, оптические и физические свойства твердофазных материалов, зависят от концентрации и энергии образования различного типа дефектов.
Если на поверхности рядом с адсорбируемым атомом находится вакансия, то возможно перемещение атомов (как адсорбированных, так и собственных атомов) по вакансионному механизму. Вероятность нахождения вакансии “рядом” тем больше, чем больше концентрация вакансий в кристалле. Поскольку энергия связи примесных и собственных атомов с атомами решетки различна, то и скорости гетеродиффузии и самодиффузии будут отличаться друг от друга. Скорость диффузии по вакансионному механизму в основном определяется концентрацией и энергией точечных дефектов.
В случае графена с одной вакансией (${\text{SV}}$) вычислили энергию образования вакансии [11]
(4)
$E_{f}^{{{\text{SV}}}} = E\left( {{\text{SV}} - {\text{graphene}}} \right) - ~\frac{{\left( {m - 1} \right)}}{m}E\left( {{\text{graphene}}} \right),$Энергии образования одиночной вакансии на графене, вычисленные с функционалами LDA и LSDA ($E_{f}^{{{\text{SV}}}} = ~$ 7.46 эВ), совпадают, и они лишь немного выше, чем энергия, полученная с функционалом PBE ($E_{f}^{{{\text{SV}}}}$ = 7.3 эВ [11]; 7.7 эВ [3]).
3.3. Зонная структура $G{{P}_{V}}\left\langle {A{{u}_{{ads}}}} \right\rangle $
Вершина валентного состояния и нижняя часть состояния проводимости чистого графена, как известно, вырождены в точках Дирака (рис. 3) зоны Бриллюэна. Вычисленная нами зонная структура 5 × 5 суперъячеек вдоль направлений высокой симметрии M-Γ-K-M на основе монослоя графена с адатомом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ представлена на рис. 4.
Валентная зона чистого графена соприкасается с соседними энергетическими зонами с нулевой запрещенной зоной ${{E}_{{\text{g}}}} = {{E}_{{\text{c}}}} - {{E}_{{\text{v}}}} \approx 0$ эВ (${{E}_{{\text{c}}}}$ – зона проводимости,$~{{E}_{{\text{v}}}}$ – валентная зона). В отсутствии примесных атомов, а также дефектов решетки, движение электронов внутри ${{E}_{{\text{g}}}}$ запрещено. Однако, расположение энергетических зон в графене похоже на расположение зон в полупроводниках и поэтому проводимость полуметалла графена хуже, чем у металлов.
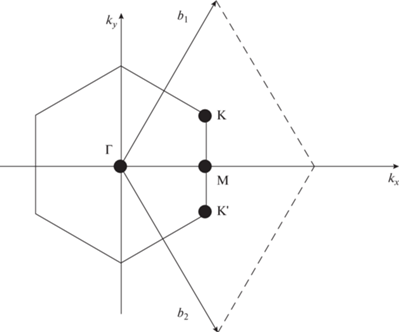
Рис. 4.
Рассчитанная электронная зонная структура 5 × 5 суперъячейки на основе графена с адатомом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ вдоль основных направлений M-Γ-K-M гексагональной зоны Бриллюэна. a – чистый графен, б – ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $, в – ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle .$ Уровень Ферми (${{E}_{{\text{F}}}}$) равен нулю. Адатом адсорбирован на Т-сайте графена.
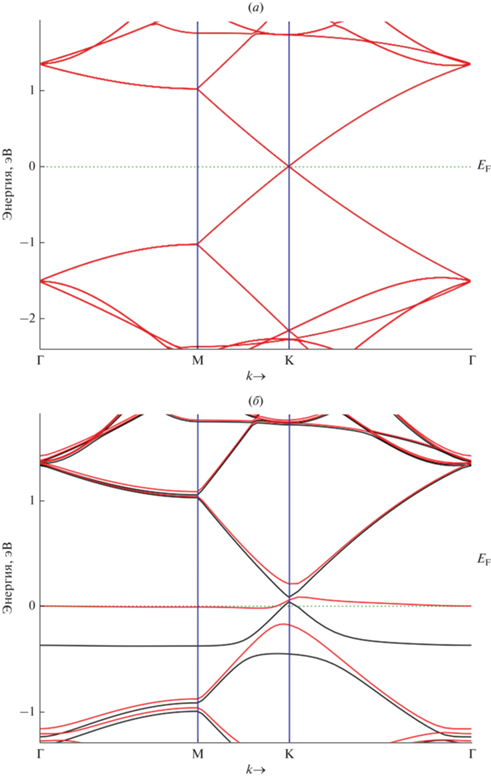
Рис. 4.
Окончание.
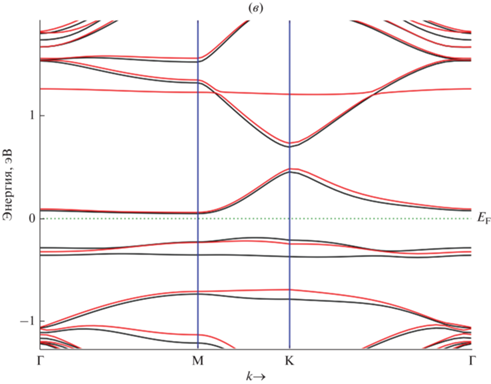
На рис. 4а показана электронная зонная структура чистого графена вдоль линии высокой симметрии зоны Бриллюэна. Монослой графена имеет полупроводниковую зонную структуру с нулевой запрещенной зоной. Как видно из рис. 4а валентная зона и зона проводимости графена соединяются в точке Дирака (в точке K-симметрии) с нулевой плотностью состояний.
Энергетические полосы валентного состояния и состояния проводимости чистого графена ${\text{GP}}$ характеризуются линейной дисперсией вблизи энергии Ферми в точке Г зоны Бриллюэна. Такая зонная структура графена (рис. 4а) приводит к нулевой эффективной массе электронов и дырок, и высокой подвижности носителей заряда в точке Дирака. Зонная структура графеновой плоскости представляет прямозонный полупроводник.
Зонная структура графена, содержащего адатом ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $, характеризуется дырочной природой проводимости (рис. 4б). Это связано с тем, что одновалентный адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ является акцептором для четырeхвалентного углерода. В результате ковалентной связи ${\text{Au}}{\kern 1pt} - {\kern 1pt} {\text{С}}$ один электрон отсутствует в одной из четырех ковалентных связей, являющихся частью решетки графена.
Аналогичное происходит также в структуре ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с участием вакансии (рис. 4в). Адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ устанавливает ковалентную связь с одним из четырех соседних атомов углерода. Для установки связи с тремя другими атомами углерода у атома золота нет валентного электрона. В этом случае он захватывает валентные электроны из ковалентной связи между соседними атомами углерода. Поэтому в структуре ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ становится отрицательно заряженным ионом и графен имеет дырочную проводимость.
Как видно из рис. 4б и в, минимум энергии валентной зоны и максимум энергии зоны проводимости графеновых структур находятся в разных точках зоны Бриллюэна. Т.е. эти структуры аналогичны непрямозонным полупроводникам. В структуре ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ формируется запрещенная зона шириной ∼0.1 эВ. Другими словами, внедрение вакансии в структуру ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ приводит к расширению запрещенной зоны ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
3.4. Плотность состояний (DOS)
На рис. 5 представлены полная и парциальные спин-поляризованные плотности состояний (PDOS) атомов золота 5 × 5 суперъячеек ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с моновакансией. Показаны плотности состояний электронов со спином соответственно вверх и вниз.
Рис. 5.
Рассчитанные полная и парциальные плотности состояний (PDOS) 5 × 5 суперъячеек графена для s-, p-, d‑электронов ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$: (а) DOS суперъячейки графена, 1 – “чистый” графен; 2 – графен с моновакансией, (б) PDOS суперъячейки ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $, (в) PDOS суперъячейки ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с моновакансией.
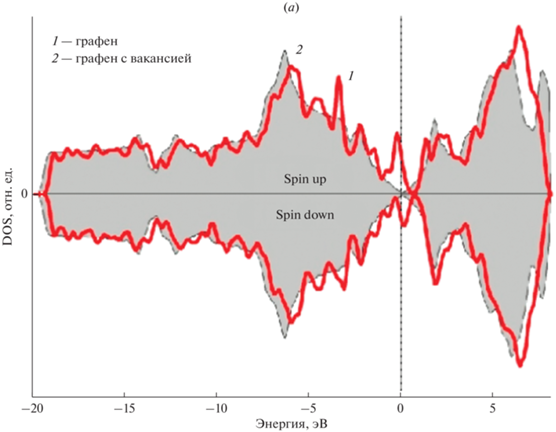
Рис. 5.
Продолжение.
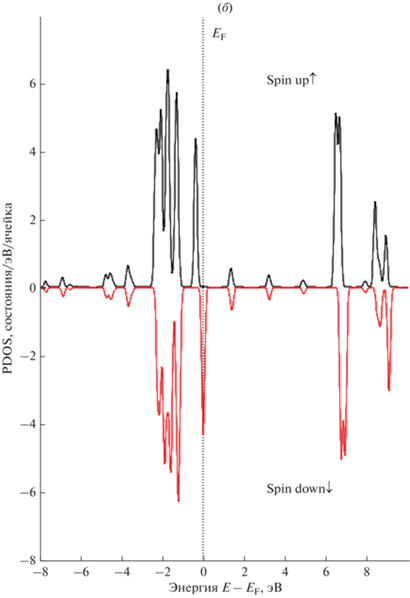
Рис. 5.
Окончание.
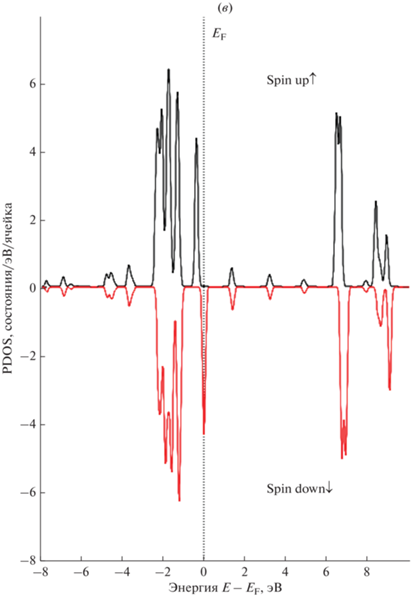
Анализ полных и парциальных спин-поляризованных плотностей электронных состояний 5 × 5 суперъячеек графена показывает, что комплекс “вакансия + адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$” на поверхности приводит к изменению DOS вблизи уровня Ферми. Заметный вклад в DOS вблизи уровня Ферми вносят d-электроны золота.
Таким образом, расположение энергетических уровней и, следовательно, плотности состояний зависят от свойств графена (например, размера и концентрации вакансии) и вклада от природы донорного атома, поставляющего свободный электрон.
3.5. Магнитный момент
Экспериментальные данные для магнитных свойств ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ отсутствуют. Для суперъячейки графена без дефектов магнитный момент (${{m}_{{{\text{cell}}}}}$ ($~{{\mu }_{{\text{B}}}}$)) должен быть равен нулю.
Магнетизм изучали методом DFT с одной вакансией углерода в суперъячейкe ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. В этом случае принимали, что вакансия изолируется и исключается взаимодействие между возможными точечными дефектами в кристалле.
Увеличение количества вакансий в GP привело бы к взаимодействию между дефектами, что усилило бы делокализацию волновой функции электронов в области недостатка углерода. Т.е. для расчета рассматривали предельный случай, где из суперъячейки ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ удаляли один атом углерода, связанный с другими атомами углерода.
Присутствие вакансии в структуре и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ приводит к возникновению магнитного упорядочения. DFT расчеты указывают на то, что в суперъячейке ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ имеет место локальное ферромагнитное спиновое упорядочение. Другими словами, система ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ на основе графена с точечным дефектом (вансия), как и аналогичные дефектные графеновые системы [11, 12], имеет локальный магнитный момент.
В табл. 2 представлены рассчитанные нами магнитные моменты для структур ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
Таблица 2.
DFT LSDA рассчитанные положение адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ на поверхности графена (${{h}_{{\text{a}}}}$), среднее расстояние от “закрепленного” атома углерода C (${{d}_{{{\text{eq}}}}}$) до ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ и общая намагниченность (${{m}_{{{\text{cell}}}}}$) адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$, адсорбированного на 5 × 5 суперъячейках ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. Данные показаны для места связывания атомов ${\text{Au}}{\kern 1pt} - {\kern 1pt} {\text{C}}$ , где адатом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ расположен сверху (T-сайт) поверхности (GP)
| Структура | ${{h}_{{\text{a}}}}({\text{{\AA}}}$) | $d_{{{\text{eq}}}}^{{\text{T}}}$$\left( {\text{{\AA}}} \right)$ | ${{m}_{{{\text{cell}}}}}$ ($~{{\mu }_{{\text{B}}}}$) |
|---|---|---|---|
| ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ | 2.59 | 2.48 | 0.91 [6] |
| ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ | 3.10 | 2.56 | 0.80 |
| ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ | 2.75 | 2.21 | 1.01 |
Таким образом, ферромагнетизм в суперъячейке ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ возникает за счет вакансии углерода и взаимодействия адатома ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ $(4{{f}^{{14}}}~4{{d}^{{10}}}~4{{s}^{1}}$) с атомами углерода. Атом золота с незаполненным уровнем вносит вклад в общую намагниченность ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $.
Для суперъячейки графена с моновакансией углерода вычисленный магнитный момент больше 1 ${{\mu }_{{\text{B}}}}$ (табл. 2). Значение ${{\mu }_{{\text{B}}}}$ для ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ отличается от значений для аналогичных структур, например, ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{G}}{{e}_{{{\text{ads}}}}}} \right\rangle $ 1.77 ${{\mu }_{{\text{B}}}}$ [12]. Высокая плотность состояний вблизи поверхности Ферми дает полный магнитный момент, что подтверждает формирование ферромагнетизма в дефектном графене ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ за счет вакансий углерода.
Установлено, что в зависимости от расстояния вакансии углерода до комплекса ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}{\kern 1pt} - {\kern 1pt} {\text{C}}$ на поверхности графена, локальный магнитный момент может как увеличиваться (в случае вакансии углерода С50), так и уменьшаться (в случае вакансии С25).
ВЫВОДЫ
DFT-LSDA расчеты 5 × 5 суперъячеек ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ показали следующие.
Расположение адсорбированных атомов золота сверху (тетраэдрическая конфигурация; T-сайт) атома углерода в поверхностном монослое графена энергетически более предпочтительно, чем в мостиковых участках между С–С связями (B-сайт) и в углублениях в центре шестичленного кольца С–С связи (H-сайт) ячейки графена.
Рассчитаны энергия образования вакансии углерода на графене ($E_{f}^{{{\text{SV}}}} = ~$ 7.46 эВ), зонная структура и плотность состояний в адсорбционных комплексах ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. Такие структуры важны для электронотранспортных свойств графеновых материалов, где носители заряда локализованы и их движение включает диффузионный процесс.
LDA-расчетные энергии адсорбции ($E_{{{\text{ads}}}}^{{\text{T}}}$) атома золота (${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$) на суперячейках графена имеют относительно низкие значения, которые характерны, например, для металлов подгруппы железа. Учет спиновой поляризации позволяет корректировать значения энергии адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ указанных графеновых структур. LSDA рассчитанное значение энергий связи с учетом спиновой поправки выше в среднем на 0.34 эВ чем LDA-расчетная $E_{{{\text{ads}}}}^{{\text{T}}}$ .
Показано, что энергия адсорбции ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ структуры ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ ниже, чем у бездефектного ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $. Энергия адсорбции в ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ и ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ зависит от дефектности поверхности и уменьшается с уменьшением степени дефектности.
Как и энергия адсорбции рассчитанные LSDA магнитные моменты (${{\mu }_{{\text{B}}}}$) для суперъячеек 5 × 5 графена с моновакансией (${\text{G}}{{{\text{P}}}_{{\text{V}}}}$) и бездефектной ячейки с адсорбированным атомом ${\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}$ отличаются друг от друга. С учетом спин-орбитального взаимодействия и расщепления f-атомных состояний ${\text{Au}}$ рассчитанный общий магнитный момент суперъячейки ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ составляет 0.8 ${{\mu }_{{\text{B}}}}$. В суперъячейке ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ с моновакансией вклад в локальный магнитный момент вносит также атом Au. В этом случае значение общего магнитного момента ${\text{G}}{{{\text{P}}}_{{\text{V}}}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ увеличивается и составляет 1.01 ${{\mu }_{{\text{B}}}}$. Магнитный момент около вакансии зависит от места расположения вакансии на поверхности. В 5 × 5 суперъячейке ${\text{GP}}\left\langle {{\text{A}}{{{\text{u}}}_{{{\text{ads}}}}}} \right\rangle $ для вакансии углерода, например, С50 и С25 значение парциального магнитного момента составляет 0.83 и 0.65${{\mu }_{{\text{B}}}}$ соответственно.
Список литературы
Zanella I., Fagan B.S., Mota R., Fazzio A. Electronic and Magnetic Properties of Ti and Fe on Graphene // The Journal of Physical Chemistry C. 2008. V. 112. № 25. P. 9163–9167. https://doi.org/10.1021/jp711691r
Ding J., Qiao Z., Feng W., Yao Y., Niu Q. Engineering quantum anomalous/valley Hall states in graphene via metal-atom adsorption: An ab-initio study // Physical Review B. 2011. V. 84. № 19. 195444–9. https://doi.org/10.1103/PhysRevB.84.195444
Singh R., Kroll P. Magnetism in graphene due to single-atom defects: dependence on the concentration and packing geometry of defects // Journal of Physics: Condensed Matter. V. 21. № 19. P. 196002–7. https://doi.org/10.1088/0953-8984/21/19/196002
Nakada K., Ishii A. Migration of adatom adsorption on graphene using DFT calculation // Solid State Communications. 2011. V. 151. № 1. P. 13–16. https://doi.org/10.1016/j.ssc.2010.10.036
Amft M., Lebègue S., Eriksson O., Skorodumova N.V. Adsorption of Cu, Ag, and Au atoms on graphene including van der Waals interactions // Journal of Physics: Condensed Matter. 2011. V. 23. № 39. P. 395001-10. https://doi.org/10.1088/0953-8984/23/39/395001
Srivastava M.K., Wang Y., Kemper A.F., Cheng H.-P. Density functional study of gold and iron clusters on perfect and defected graphene // Physical Review B. 2012. V. 85. № 16. P. 165444-13. https://doi.org/10.1103/PhysRevB.85.165444
Castillo R.M.D., Sansores L.E. Study of the electronic structure of Ag, Au, Pt and Pd clusters adsorption on graphene and their effect on conductivity // The European Physical Journal B. 2015. V. 88. № 248. P. 1–13. https://doi.org/10.1140/epjb/e2015-60001-2
Trentino A., Mizohata K., Zagler G., Längle M., Mustonen K., Susi T., Kotakoski J., Åhlgren E.H. Two-step implantation of gold into graphene // 2D Materials. 2022. V. 9. № 025011. https://doi.org/10.1088/2053-1583/ac4e9c
Engel J., Francis S., Roldan A. The influence of support materials on the structural and electronic properties of gold nanoparticles – a DFT study // Physical Chemistry Chemical Physics. 2019. rsc.li/PCCP. https://doi.org/10.1039/c9cp03066b
Plant S.R., Cao L., Yin F., Wang Z.W., Palmer R.E. Size-dependent propagation of Au nanoclusters through few-layer graphene // Nanoscale. 2014. V. 6. P. 1258–1268. https://doi.org/10.1039/c3nr04770a
Асадов M.M., Мустафаева С.Н., Гусейнова С.C., Лукичев В.Ф., Тагиев Д.Б. Ab initio моделирование влияния расположения и свойств упорядоченных вакансий на магнитное состояние монослоя графена // Физика твердого тела. 2021. Т. 63. № 5. С. 680–689. [Asadov M.M., Mustafaeva S.N., Guseinova S.S., Lukichev V.F., Tagiev D.B. Ab initio modeling of the location and properties of ordered vacancies on the magnetic state of a graphene monolayer // Physics of the Solid State. 2021. V. 63. № 5. P. 680–689.] https://doi.org/10.1134/S1063783421050036
Асадов М.М., Мустафаева С.Н., Гусейнова С.С., Лукичев В.Ф. DFT моделирование электронной структуры и адсорбция германия в упорядоченном графене с вакансией // Микроэлектроника. 2022. Т. 51. № 2. С. 125–139. [Asadov M.M., Mustafaeva S.N., Guseinova S.S., Lukichev V.F. DFT Electronic Structure Simulation and Adsorption of Germanium in Ordered Graphene with a Vacancy. Russian Microelectronics. 2022. V. 51. № 2. P. 83–96.] https://doi.org/10.1134/S1063739722010024
Perdew J.P., Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Physical Review B. 1981. V. 23. № 10. P. 5048–5079. https://doi.org/10.1103/physrevb.23.5048
Monkhorst H.J., Pack J.D. Special points for Brillouin-zone integrations. Physical Review B. 1976. V. 13. № 12. P. 5188–5192. https://doi.org/10.1103/physrevb.13.5188
Arthur J.R., Cho A.Y. Adsorption and desorption kinetics of Cu and Au on (0001) graphite. Surface Science. 1973. V. 36. № 2. P. 641–660. https://doi.org/10.1016/0039-6028(73)90409-3
Anton R., Kreutzer P. In situ TEM evaluation of the growth kinetics of Au particles on highly oriented pyrolithic graphite at elevated temperatures. Physical Review B. 2000. V. 61. № 23. P. 16077–16083. https://doi.org/10.1103/PhysRevB.61.16077
Дополнительные материалы отсутствуют.
Инструменты
Микроэлектроника