

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 1, стр. 29-42
ОСОБЕННОСТИ ТЕРМОДЕСТРУКЦИИ НАПОЛНЕННЫХ КОМПОЗИЦИЙ НА ОСНОВЕ БУТАДИЕН-СТИРОЛЬНОГО ТЕРМОЭЛАСТОПЛАСТА
Л. С. Шибряева a, b, *, Н. Д. Блинов a, Л. Р. Люсова b, Ю. А. Наумова b
a Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
119334 Москва, ул. Косыгина, 4, Россия
b МИРЭА/ИТХТ – Российский технологический университет/
Институт тонких химических технологий имени М.В. Ломоносова
119435 Москва, ул. Малая Пироговская, 1, стр. 5, Россия
* E-mail: lyudmila.shibryaeva@yandex.ru
Поступила в редакцию 22.10.2021
После доработки 12.11.2021
Принята к публикации 15.12.2021
- EDN: KUSRTW
- DOI: 10.31857/S2308113922020048
Аннотация
Изучены особенности структуры и процесса термодеструкции композиций из бутадиен-стирольного термоэластопласта с биодеградируемым полимером полилактидом и поверхностно-активными веществами: тетраметилендиэтилентетрамином и дистеарилдиметиламмоний хлоридом. С помощью растровой электронной микроскопии установлены особенности морфологии композиций, а методом термогравиметрии и дифференциальной термогравиметрии определены термические и кинетические параметры разложения компонентов композиции. Кинетические параметры термодеструкции рассчитывали с использованием моделей Freeman–Carroll, Friedmen–Ozawa, Coats–Redfern. При рассчете параметров деструкции применяли метод разделения дифференциально-термической кривой на отдельные пики, характеризующие индивидуальные компоненты. Показано, что введение полилактида и поверхностно-активных веществ в бутадиен-стирольный термоэластопласт приводит к изменению кинетики и механизма терморазложения полибутадиенового и полистирольного компонентов. Полилактид увеличивает, а поверхностно-активные вещества уменьшают максимальную скорость потери массы композиции по сравнению с индивидуальным термоэластопластом. Влияние наполнителей на кинетику термодеструкции обусловлено их межмолекулярным взаимодействием с термоэластопластом. Показано, что особенности термодеструкции наполненного бутадиен-стирольного термоэластопласта определяются локализацией полилактида в бутадиен-стирольной матрице вокруг доменов полистирола, а поверхностно-активных веществ – в полилактиде и матрице бутадиен-стирольного термоэластопласта, включая блоки полистирола.
ВВЕДЕНИЕ
Проблема поиска новых биодеградируемых материалов, стойких к высокотемпературному воздействию и термодеструкции остается актуальной. Это связано прежде всего с появлением новых областей применения полимеров – в медицине, фармакологии, пищевой промышленности [1–12]. В частности, биосовместимые полимеры используют при создании материалов для изготовления медицинских изделий и оборудования, в том числе имплантатов для суставов, кровеносных сосудов, сердечных клапанов, при получении пролонгированных лекарственных форм, раневых покрытий, упаковок, диагностических инструментов и т.д. [4–9, 12]. При получении, переработке и эксплуатации биосовместимых полимеров и композиционных материалов на их основе необходимо решать проблему стабилизации таких полимерных систем [13–16].
Для изделий медицинского назначения также требуется создание материалов, обладающих термостойкостью и антибактериальными свойствами. Технологией создания такого материала является модификация поверхностного слоя полимера, из которого изготовлен медицинский инструментарий. Наиболее подходящим полимером, поверхностный слой которого может быть подвержен модификации с целью обеспечения требуемых свойств, служит бутадиен-стирольный термоэластопласт (ДСТ). Введение в эластомерную основу биоразлагаемых пластиков совместно с поверхностно-активными веществами позволяет достичь высоких антибактериальных свойств [9, 17, 18]. Введение в ДСТ наполнителей различной природы может изменить физико-химические свойства, кинетику и механизм термодеструкции термоэластопласта и изделий из него [19–21]. Использование медицинского оборудования предполагает его высокотемпературную стерилизацию, в связи с чем вопрос характера влияния наполнителей на термостабильность бутадиен-стирольной матрицы очень важен.
Анализ литературы показал существование разных и порой противоречивых сведений о характере влияния наполнителей и модификаторов на термостойкость полимерных композиций [21, 22]. Разный характер влияния наполнителей на терморазложение полимера обусловлен многими причинами. Например, присутствие модификатора может вызвать ингибирование процесса деструкции ДСТ или создать ограничения сегментарной подвижности цепей в матрице, вследствие чего уменьшится скорость разрушения композиции [22, 23].
Основными факторами, определяющими термостабильность композиции, являются ее состав, природа компонентов и структура [13–16, 19–23]. Морфология композиции в значительной степени облегчает создание материалов с заданными свойствами, а также может носить предсказательный характер. В связи с этим задача интерпретации зависимостей состав–структура–термостойкость на примере модифицированного ДСТ актуальна и имеет практическое значение.
Цель настоящей работы – установление особенностей механизма и кинетики термодеструкции композиций из бутадиен-стирольного термоэластопласта, наполненного биодеградируемым полилактидом совместно с поверхностно активными веществами, и выявление факторов, определяющих эти особенности.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали следующие соединения.
Бутадиен-стирольный термоэластопласт ДСТ-30-01 производства Открытого акционерного общества “Воронежсинтезкаучук” по ТУ 38.103.267-99 в виде гранул; содержание связанного стирола в блок-сополимере составляет 27–31 мас. %; показатель текучести расплава 1 г/10 мин (при 190°С, 5 кгс); содержание летучих веществ 0.5 мас. %, золы 2.0 мас. %, стабилизатора 0.2–0.5 мас. %; условная прочность при растяжении – не менее 19.6 МПа, относительное удлинение при разрыве – не менее 650%.
Полилактид (ПЛА), L-, D,L-энантиомер Ingeo Biopolymer 4032D в виде гранул, с молекулярной массой 1.7 × 105, плотностью 1.27 г/см3 , показатель текучести расплава 4 г/10 мин (при 185°С, 2.5 кгс), пределом текучести при растяжении 60 МПа, относительным удлинением при разрыве 6%, температурой плавления 170°С, степенью кристалличности 50–60%.
Тетраметилендиэтилентетрамин (ТМДЭТА, теотропин, (1,8,3,6-диэндометилен-1,3,6,8-тетраазациклодекан), циклический третичный амин, синтезируемый конденсацией этилендиамина с формальдегидом, антибактериальный агент с противовирусной активностью – желтовато-белый порошок, легко растворимый в воде, спиртах, хлороформе, Тпл = 195°С.
Дистеарилдиметиламмония хлорид (ДСДМАХ, талофлок (N,N-диметил-N,N-диоктадециламмония хлорид) – четвертичная соль аммония, белый порошок плотностью 0.84 г/см3, не растворимый в воде, растворимый в спиртах, кетонах, температура разложения 135°С, при хранении стабилен, в том числе в виде спиртовых растворов; катионный ПАВ, антибактериальный агент широкого спектра действия.
Композиции, содержащие ДСТ, ПЛА, ТМДЭТА и ДСДМАХ, изготавливали путем растворения компонентов в хлороформе; пленки получали испарением растворителя.
Растровую электронную микроскопию (РЭМ) материала проводили в Центре коллективного пользования научными приборами на базе Московского государственного технического университета имени Н.Э. Баумана. Для испытания использовали образцы пленок размером 1 × 1 см и толщиной 100 мкм.
Процесс термодеструкции образцов изучали с помощью термогравиметрического анализа на термомикровесах TG 209 F1 Iris (“Netzsch”) в динамических условиях нагревания в токе аргона. Для испытания изготавливали образцы пленок в форме диска. Навески образцов составляли 5–8 мг, анализ изменения массы образца в зависимости от температуры проводили при скорости нагревания 20 град/мин. В случае термического разложения образцы нагревали в токе аргона при скорости потока 100 см–3/мин. Также были получены дифференциальные кривые (ДТГА); точность определения температур ±2°С.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Особенности структуры композиции
Как известно, ПЛА термодинамически несовместим с ДСТ [24], однако в образцах с невысокой концентрацией ПЛА (5–10%), полученных с помощью растворной технологии, проявляется взаимодействие компонентов ДСТ и ПЛА, при этом отмечено влияние ПЛА на структуру бутадиен-стирольного термоэластопласта. Важно отметить, что параметр растворимости ПЛА (δ = = 20.3 (МДж/м3)1/2) ближе к параметру растворимости полистирола, составляющего жесткие блоки ДСТ (δ = 18.2), чем к параметру растворимости эластомерной фазы полибутадиена (δ = 16.9). При испарении растворителя из раствора ДСТ/ПЛА происходит определенное распределение частиц ПЛА в матрице ДСТ. Исходя из значений параметров растворимости, можно предположить, что частицы ПЛА должны находиться в полистирольной фазе ДСТ. Но при испарении растворителя ПС переходит в стеклообразное состояние, поэтому он должен вытеснять частицы ПЛА на периферию растущего домена. Такая картина распределения обнаруживается с помощью электронной микроскопии.
Для изучения структуры исследуемых материалов на основе ДСТ был использован метод растровой электронной микроскопии (РЭМ), где для получения изображения использовались сигналы, образованные вторичными электронами. На рис. 1 представлены РЭМ-изображения поверхности пленок из ДСТ и ДСТ + 10% ПЛА. Светлые области, обладающие большей плотностью, соответствуют доменам ПС, темные – эластомерной фазе ПБ. На рис. 1 видно, что введение ПЛА в ДСТ приводит к изменению в структуре поверхности пленочного образца. На РЭМ-изображении ПЛА представлен в виде светлых колец. Такие выводы были сделаны на основании анализа электронной картины, которая постоянно изменялась, кольца находились в движении. Это связано с тем, что при взаимодействии электронов с ПЛА происходит его разложение мощной эмиссией вторичных электронов, высвечивающих области разложения. Таким образом, кольцеобразные структуры соответствуют ПЛА, который локализуется в эластомерной фазе вокруг более темных областей, соответствующих доменам ПС. Повышение контрастности снимка из-за вторичной эмиссии при разложении ПЛА не позволяет сравнить в данном случае электронную плотность ПС и ПБ. Тем не менее, если бы ПЛА находился в неупорядочном состоянии в эластомерной фазе, наблюдаемая картина не содержала бы кольцеобразных структур. Введение ТМДЭТА или ДСДМАХ от 0.5 до 2.5 мас. % существенно не влияет на характер поверхности на изображениях РЭМ.
Рис. 1.
РЭМ-изображения поверхности пленок из ДСТ (а) и композиции ДСТ + 10% ПЛА (б). Цветные рисунки можно посмотреть в электронной версии.

Таким образом, данные РЭМ свидетельствуют о локализации ПЛА в массе эластомерного компонента ДСТ вокруг доменов ПС на границе ПБ–ПС.
Формальная кинетика термодеструкции композиций
Для изучения особенностей процесса термодеструкции композиций из ДСТ с наполнителями был использован метод термогравиметрического анализа. Термограммы регистрировали с помощью динамического ТГА в режиме постоянной скорости сканирования в инертной среде (аргон) и дифференциированием кривых ТГА получали ДТГА. На всех кривых ДТГА видны отчетливые пики, характеристика которых представлена ниже (пики пронумерованы в порядке повышения температуры).
Пик 1 (150–175°С) появляется только в случае, если в композиции есть ТМДЭТА. Его наличие может быть связано как с разложением ТМДЭТА на этилендиамин и формальдегид, так и с его сублимацией.
Пик 2 наблюдается в интервале температур 250–380°С в индивидуальном ПЛА и при его наличии в составе композиции (рис. 2б–2г). Следует отметить, что температура отрыва групп >C=O в ПЛА составляет 270°С, что соответствует пику деструкции чистого ПЛА (кривая 2), поэтому пик 2 в композициях однозначно может быть отнесен к ПЛА .
Рис. 2.
Кривые ТГА (штриховые линии) и ДТГА (сплошные): а – ДСТ(1', пик 2) и ПЛА (2', пики 3–6); б − ДСТ + 10% ПЛА; в − ДСТ + 10% ПЛА + 2.5% ТМДЭТА; г − ДСТ+ 10% ПЛА + 2.5% ДСДМАХ.
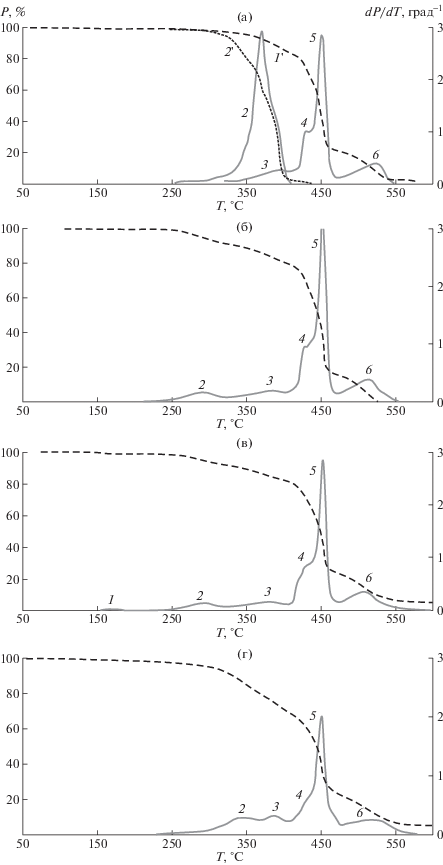
Наличие пиков 3–6 на всех графиках позволяет сделать вывод об их принадлежности к ДСТ.
Пик 3 (380°С) является самым стабильным, он не смещается и почти не изменяется в разных композициях. Можно предположить, что данный пик связан с выделением стирола-мономера, причем статистически присоединенного к ПБ (который обязательно присутствует в ДСТ), и разложением стирол-бутадиенового статистического сополимера, преобладающего у границ ПС-блоков макромолекул ДСТ и содержащего преимущественно структуру 1,2-ПБ.
Пик 4 (420°С) представляет собой левое плечо самого большого пика 5. Учитывая принадлежность пика 5 к структуре транс-1,4-ПБ (см. ниже), и то, что температура деструкции цис-изомеров ниже, чем транс-изомеров, пик 4 следует отнести к структуре цис-1,4-ПБ.
Пик 5 (450°С) наиболее интенсивен и потому относится к структуре 1,4-транс-ПБ, поскольку именно она является преобладающей в ДСТ.
Пик 6 с максимумом при 520°С соответствует разложению ПС-блоков как самых термостойких. Уширение пика от 550 к 580°С обусловлено большим количеством конкурирующих процессов разложения.
При сравнении данных ТГА/ДТГА, полученных для ДСТ, ПЛА и наполненных образцов можно отметить влияние наполнителей на положение на шкале температур пиков, отвечающих за разложение компонентов, изменение в величинах максимальных температур Tмакс и максимальных скоростях потери массы (dP/dT). Прежде всего из данных ТГА следует, что при введении в индивидуальный ДСТ 10% ПЛА температура 5%-ной потери массы, уменьшается с 355.55°С до 298.75°С. При этом температура 5%-ной потери массы индивидуального ПЛА составляет 327.5°С. В то же время введение в композицию ДСТ–ПЛА наполнителей ТМДЭТА и ДСДМАХ мало влияет на величину указанных температур, они равны 289.2 и 297.6°С соответственно.
На кривых ДТГА видно, что смешение ДСТ с ПЛА (рис. 2а, 2б) приводит к следующим изменениям. Пик 2, характеризующий распад индивидуального ПЛА, для смеси ДСТ–ПЛА смещается в сторону более низкой температуры (рис. 2а кривая 2 ' и рис. 2б–2г). Его максимальная температура разложения Тмакс понижается с 355 до 290°С. Аналогичным образом во всех композициях ДСТ/ПЛА с ПАВ пик 2 ПЛА проявляется при более низкой температуре (табл. 1), 294.1 и 339.3°С для композиций ДСТ–ПЛА–ТМДЭТА и ДСТ–ПЛА–ДСДМАХ соответственно. Смещение пика 2 в низкотемпературную область указывает на ускорение начала термодеструкции полимера, по всей видимости, за счет распада компонента ПЛА. С введением ПЛА в ДСТ увеличивается интенсивность пика 5, относящегося к распаду 1,4-транс-ПБ в ДСТ, что свидетельствует о росте максимальной скорости процесса. Также наблюдается тенденция к смещению от 525 до 510°С Тмакс пика 6, характеризующего реакции распада ПС-блоков в ДСТ.
Таблица 1.
Термофизические параметры деструкции наполненных композиций ДСТ, полученные из кривых ТГА и ДТГА
| Образец | Пик | Температурный интервал пика, Тк – Тн, °С | Максимальная температура пика, Тмакс °С | Величина потери массы (степень превращения, соответствующая Тмакс), dP/dT |
|---|---|---|---|---|
| ПЛА | 2 | 280–425 | 355 | 100 |
| ДСТ | 3 | 300–421 | 391 | 10 |
| 4 | 421–435 | 429 | 32 | |
| 5 | 435–465 | 450 | 95 | |
| 6 | 465–550 | 525 | 14 | |
| ДСТ + 10% ПЛА | 2 | 229–321 | 290 | 7 |
| 3 | 321–413 | 387 | 7.5 | |
| 4 | 413–434 | 429 | 32 | |
| 5 | 434–471 | 452 | 100 | |
| 6 | 471–560 | 510 | 15 | |
| ДСТ + 10% ПЛА + 2.5% ТМДЭТА | 1 | 145–180 | 165 | 2 |
| 2 | 203–327 | 294 | 4.8 | |
| 3 | 327–405 | 382 | 5 | |
| 4 | 411–437 | 429 | 25 | |
| 5 | 437–474 | 450 | 93 | |
| 6 | 475–580 | 509 | 15 | |
| ДСТ + 10% ПЛА + 2.5% ДСДМАХ | 2 | 235–400 | 340 | 10 |
| 3 | 367–405 | 385 | 10 | |
| 4 | 408–432 | 426 | 17 | |
| 5 | 432–472 | 450 | 68 | |
| 6 | 472–580 | 515 | 7 |
С одной стороны, смещение пика 2 деструкции ПЛА и пика 6 ДСТ в низкотемпературную область, указывающее на ускорение деструкции может быть обусловлено взаимодействием ПЛА с блоками ПС. С другой стороны, смещение пика 2 и рост интенсивности пика 5, т.е. максимальной скорости распада, указывает на инициирование процесса деструкции ПБ продуктами распада ПЛА. Учитывая термодинамическую несовместимость ПЛА и ПС, низкотемпературное смещение пика 2, а также рост интенсивности пика 5, относящегося к связям транс-1,4-ПБ, примыкающим к границам ПС-блоков, можно предположить распределение частиц ПЛА в менее жесткой эластомерной фазе, а именно, в среде статистического бутадиен-стирольного сополимера у границ ПС-блоков. Такая локализация ПЛА создает благоприятные условия для инициирования распада эластомера. Этот факт подтверждают данные РЭМ, согласно которым ПЛА распределяется в ДСТ преимущественно у блоков ПС.
Введении ТМДЭТА в ДСТ приводит к появлению на кривой ДТГА (рис. 2в ) пика 1, очевидно, относящегося к фазовому переходу ТМДЭТА. Появление этого пика соответствует уменьшению пиков 3–6. Интенсивность пика 3, отвечающего максимальной скорости разложения структур 1,2-ПБ в макромолекулах ДСТ, понижается с величины, характерной для чистого ДСТ, равной 0.27, до 0.18 град–1 в композициях ДСТ–ПЛА, ДСТ–ПЛА–ТМДЭТА (табл. 1). В то же время она увеличивается в композиции ДСТ–ПЛА–ДСДМАХ (табл. 1) до 0.32 град–1.
Максимальная скорость распада 1,4-транс-ПБ в ДСТ (пик 5) равна 2.8 град–1 в индивидуальном ДСТ, увеличиваясь до 3.0 в композиции ДСТ/ПЛА, но уменьшается до 2.8 и 2.0 град–1 в композициях ДСТ–ПЛА–ТМДЭТА и ДСТ–ПЛА–ДСДМАХ соответственно (табл. 1).
Это, по-видимому, свидетельствует о замедлении терморазложения ДСТ, проявлении стабилизирующего действия ТМДЭТА на распад связей ПБ в ДСТ. Изменения пика 2 при этом практически не заметно. Однако при добавлении ДСДМАХ (рис. 2г) происходит смещение указанного пика в сторону высоких температур, благодаря чему его интенсивность возрастает. Несмотря на то, что пик 3 почти не смещается по температуре, его интенсивность также увеличивается. При этом в системе наблюдается тенденция к уменьшению максимальной скорости распада блоков ПС (пик 6). Значение dP/dT = 0.39 в чистом ДСТ уменьшается до 0.27 град−1 в системе с ДСДМАХ. Данные изменения можно объяснить тем, что ДСДМАХ стабилизирует ПЛА, причем при повышении температуры стабилизирующее действие понижается, тем самым вызывая интенсивное начало разложения полимера: левое плечо 4 большого пика 5 стало малозаметным, а интенсивность пика 5 значительно уменьшилась. Понижение интенсивности этого пика, свидетельствующее о замедлении разложения эластомерного компонента ДСТ и может быть связано со стабилизирующим действием ДСДМАХ на ПБ-блоки. Предположительно, ДСДМАХ стабилизирует ПС (пик 6 становится ниже), причем ДСДМАХ, находясь и внутри блоков ПС, меняет характер деструкции и увеличивает количество реакций разложения, что проявляется в уширении пика 6 (рис. 2г). Итак, можно считать, что ДСДМАХ стабилизирует ПЛА, увеличивает температуру его разложения и стабилизирует ДСТ, понижая скорость деструкции, распределяясь как в ПБ-, так и в ПС-блоках.
Механизмы термодеструкции композиций
Анализ терморазложения композиций исходит из положения, что процесс может быть представлен следующей схемой:
В качестве параметров, характеризующих механизм процесса, использовали энергию активации Ea, предэкспоненциальный фактор A и показатель порядка реакции n.
Для расчета указанных параметров были применены кинетические модели [25–33], в основе которых лежит температурная зависимость скорости процесса k(T) и конверсионной составля-ющей f(α).
Скорость превращения полимера может быть представлена в виде произведения этих функций:
где α – конверсия или степень изменения полимера в ходе реакции.Для метода ТГА величина α определяется уравнением
в котором m0 – начальное значение массы образца выбранного для анализа диапазона данных, m∞ – конечное значение массы образца выбранного диапазона данных, mτ – масса образца в момент времени τ или равная отношению веса полимера, который улетучился к исходному весу, $v$ – скорость изменения конверсии α или состава полимера за единицу времени t.Разработанные на сегодняшний день методы получения кинетических параметров включают в себя анализ либо одной термограммы, полученной в условиях постоянной скорости изменения температуры образца β, либо нескольких термограмм с различными постоянными скоростями нагревания. В настоящей работе был применен неизотермический режим термодеструкции при одной постоянной скорости нагревания образца. Для расчета параметров термодеструкции изученных композиций такой подход был выбран с целью минимизирования ошибки эксперимента, обусловленной неоднородностью распределения компонентов, подверженных термовоздействию в условиях разных скоростей нагревания.
При проведении эксперимента в неизотермических условиях при постоянной скорости нагревания образца β = dT/dt скорость изменения конверсии может быть представлена соотношением
где k(T) – температурная зависимость скорости потери массы, описывается уравнением Аррениуса Здесь E – эффективная энергия активации, A – предэкспоненциальный фактор, R – универсальная газовая постоянная.Конверсионно-зависимую функцию f(α) для потери массы, можно представить в виде
соотношение (1 – α) может быть заменено на W – весовую фракцию остающуюся в ходе изменения веса на термограмме ТГА или в дифференциальном виде(5)
${\text{ln}}{v} = {\text{ln}}\left( { - \frac{{dW}}{{dt}}} \right) = {\text{ln}}A + n~{\text{ln}}W - \frac{E}{{RT}}$В работе были использованы различные модельные методы, направленные на получение линеаризованных графиков зависимостей данных от обратных температур, для обеспечения быстрой визуальной оценки порядка реакции, ее энергии активации и предэкспоненты.
Метод Freeman–Carroll. В дифференциальной форме уравнение (5) выглядит следующим образом:
(6)
$\Delta {\text{ln}}\left( { - \frac{{dW}}{{dt}}} \right) = n~\Delta ~{\text{ln}}W - \left( {\frac{E}{R}} \right)\Delta \left( {\frac{1}{T}} \right)$Или в преобразованном виде:
(7)
$\left( {\frac{{\Delta {\text{ln}}\left( {\frac{{dw}}{{dt}}} \right)}}{{\Delta \left( {\frac{1}{T}} \right)}}} \right) = n~\left( {\frac{{\Delta {\text{ln}}W}}{{\Delta \left( {\frac{1}{T}} \right)}}} \right) - \left( {\frac{E}{R}} \right)$(8)
$\left( {\frac{{\Delta {\text{ln}}\left( {\frac{{dW}}{{dt}}} \right)}}{{\Delta {\text{ln}}W}}} \right) = n + E\left( { - \frac{{\Delta \left( {\frac{1}{T}} \right)}}{{R\Delta {\text{ln}}W}}} \right)$Отсюда вытекают две графические зависимости:
(9)
$[\Delta {\text{ln}}\left( {dW{\text{/}}dt} \right){\text{/}}\Delta \left( {1{\text{/}}T} \right)] = f[\Delta {\text{ln}}W{\text{/}}\Delta \left( {1{\text{/}}T} \right)]~$(10)
$[\Delta {\text{ln}}\left( {dW{\text{/}}dt} \right){\text{/}}\Delta {\text{ln}}W] = f[ - \Delta \left( {1{\text{/}}T} \right){\text{/}}R\Delta {\text{ln}}W]$Зависимости в координатах (9) и (10) представляют собой прямую линию. В первом случае ее наклон равен порядку химической реакции, а отсечение ординаты при нулевом значении на оси абсцисс величине параметра (–Eа/R). В случае зависимости (10) наклон прямой линии равен Eа, а отсечение – величине n.
Метод Friedman–Ozawa. Этот метод используют в случае кинетической зависимости n-го порядка. Термокинетическая модель для одной постоянной скорости нагревания может быть представлена следующим уравнением:
(11)
$\left( {{\text{ln}}\frac{{dW}}{{dt}} - n~{\text{ln}}W} \right) = E~\left( { - \frac{1}{{RT}}} \right) + \ln A$При n ~1 ${\text{ln}}\left( {\frac{{dW{\text{/}}dt}}{W}} \right) = E~\left( { - \frac{1}{{RT}}} \right) + {\text{ln}}A$ получаем соотношение
(12)
${\text{ln}}\left[ {--\left( {dW{\text{/}}dt} \right){\text{/}}W} \right] = f\left( {--1{\text{/}}RT} \right)~$Для постоянной Eа в определенном температурном интервале, данная зависимость имеет линейный характер, наклон которой соответствует E. Отсечение на оси ординат при нулевом значении оси абсцисс составляет ln A.
Появление перегибов на указанной линейной зависимости может быть связано с переходом псевдопервого порядка реакции на другой или изменением величины энергии активации в связи со сменой механизма процесса.
Метод Coats–Redfern. В данном методе используется уравнение
В работе значения Еа определяли из наклонов прямых на графиках зависимостей в координатах:
Известно, что трудности термического анализа возникают в тех случаях, когда различные процессы, связанные с выделением или поглощением энергии, протекают одновременно и накладываются друг на друга. Так, эндотермический эффект деструкции полимерных цепей может накладываться на эндотермический эффект сублимации или испарения продуктов деструкции [34].
Одновременное протекание таких процессов может приводить к возникновению сингулярностей на термограммах.
При исследовании механизма термодеструкции композитов ДСТ, наполненных ПЛА, ТМДЭТА и ДСДМАХ учитывали, что брутто-процесс термоокислительной деструкции как ДСТ, так и композиции складывается из нескольких независимых эндотермических реакций. При этом распределение температуры в пределах образца единообразно, время потери тепла линейно зависит от температуры в разных фракциях полимерного вещества, т.е. в реактантах 1, 2, 3 … j. Тогда пользуясь моделями, предложенными в работах [35, 36], брутто-процесс деструкции можно представить следующей схемой:
Здесь в композиции ДСТ с наполнителями, [RH] – суммарная концентрация реакционноспособных активных центров, участвующих в процессе деструкции; [RH1]–[RH6] – концентрация активных связей компонентов материала, включая ПЛА, отдельные компоненты, составляющие ДСТ, ТМДЭТА, ДСДМАХ; θ1−6 − кинетические параметры реакций распада связей; t – время распада.
Композиция наполненного ДСТ − система гетерогенная, кинетические уравнения включают параметры энергии активации и предэкспоненты, не являющиеся постоянными величинами [37]. В системах существует обмен свободной валентностью между различными активными центрами, осуществляется межмолекулярная передача кинетических цепей окисления и деструкции. Высокомолекулярная природа и особенности структуры создают ограничение для перераспределения энергии между активными центрами и реакционноспособными функциональными группами. В результате возникает распределение реакционных центров по кинетическим параметрам.
Для выявления особенностей механизма термодеструкции изучаемых композиций на основе ДСТ и установления изменений в процессе, к которым приводит введение наполнителей, были проанализированы кривые температурных зависимостей, полученных с помощью различных моделей для каждого из пиков 1–6, относящихся к разным компонентам (фракциям) в ДСТ и композициям ДСТ с наполнителями. Эти пики выделены из кривых ДТГА. Задачу определения температурного интервала, вводимого в уравнение теплового баланса, решали методом Тихонова [38], считая что каждый пик описывается Гауссовой формой. На рис. 3 представлен образец разделения пиков на примере ДСТ и ДСТ с ПЛА. Для выделенных пиков, были получены кинетические параметры: величины α, Еаj и Аj, характеризующие каждую фракцию композиции, которая участвует в реакции соответственно своей локальной структуре, описывает процесс разрушения связей, отличающихся своей термостабильностью, его механизм и вносят соответствующий вклад в брутто-процесс. Соответственно количество фракций и их параметры различаются в зависимости от природы наполнителя и структуры образца. С целью установления порядка реакции для каждого из пиков на кривой ДТГА исследуемых образцов применяли кривые полулогарифмических зависимостей, полученных с помощью модели Freeman–Carroll в кординатах (9) и (10). Примеры зависимостей в координатах [(Δln(dW/dt))/(ΔlnW)] − f[−Δ(1/T)/R(ΔlnW)] для образца ПЛА (пик 2), в координатах [Δln(dW/dt)/Δ(1/T)] − f [ΔlnW/Δ(1/T)] для ДСТ (пик 3) и для смеси состава ДСТ + 10% ПЛА (пик 5) представлены на рис. 4.
Рис. 3.
Область кривой ДТГА для ДСТ и ДСТ + 10% ПЛА, на которой представлено выделение отдельных пиков 2–6. Пояснения в тексте.
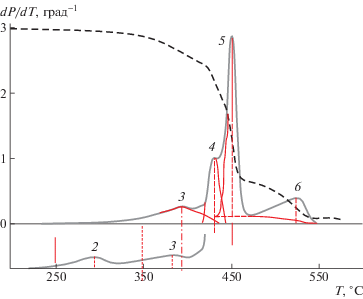
Рис. 4.
Полулогарифмические температурные зависимости скоростей превращения полимеров в ходе деструкции, представленные моделями Freeman–Carroll в координатах Δln(dW/dt)/Δ(1/T)−fΔlnW/Δ(1/T) для образца ДСТ в области температур пика 3 (а), а также в координатах [(Δln(dW/dt)/(ΔlnW)]−f[−Δ(1/T)/ R(ΔlnW)] для ПЛА в области температур пика 2 (б) и композиции ДСТ + 10% ПЛА в области 425–468оС пика 5 (в).
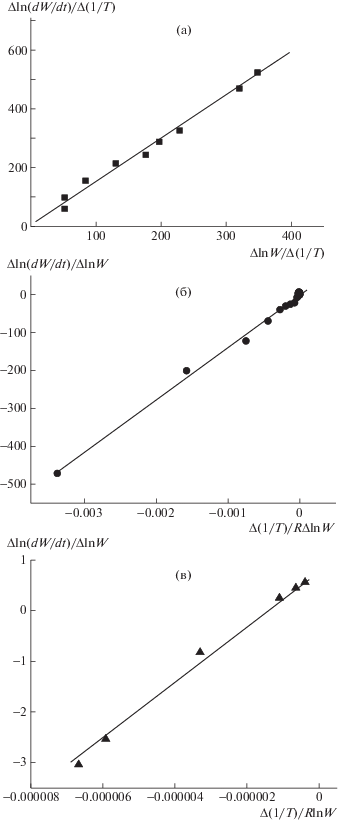
Значения величин n и Еа, полученные из указанных соотношений для каждой фракции, приведены в табл. 2. Как видно, величины n всех изученных образцов близки к единице. Для определения эффективной энергии активации и предэкспоненты была использована модель Friedman–Ozawa применительно к неизотермическому режиму с одной скоростью нагревания образца в соответствии с уравнением (11). Для более точного определения параметров деструкции также была привлечена модель Coats–Redfern с использованием соотношения (13). Для каждого из пиков 1–6 всех изученных образцов были получены линеаризованные кривые с применением трех моделей. Из них определены кинетические параметры деструкции Ea и A. Указанные параметры, полученные по разным моделям, удовлетворительно совпадают (табл. 2). Анализ этих данных показал существование перегибов на линеаризованных кривых, связанных с температурными интервалами не только характерными для разных пиков, но и внутри одного пика, что свидетельствует об изменении параметров процесса разрушения полимеров.
Таблица 2.
Значения показателя порядка реакции, эффективной энергии активации, предэкспоненты процесса термодеструкции ДСТ и композиций на его основе, полученные с применением трех моделей
| Образец | Пик | Уравнение Freeman–Carroll | Уравнение Friedman–Ozava | Уравнение Coats–Redfern | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔТ, °C | Δα, % | Ea, кДж/моль | n | ΔТ, °C | Δα, % | Ea, кДж/моль | A, c–1 | ΔТ, °C | Δα, % | Ea, кДж/моль | ||
| ПЛА | 2 | 262–400 | 2–91 | 138 ± 10 | 1.2 | 262–400 | 2–91 | 144 ± 10 | 1010–11 | 262–400 | 2–91 | 154 ± 10 |
| ДСТ | 3 | 325–420 | 6.5–70 | 116 ± 10 | 1.5 | 325–420 | 6.5–70 | 112 ± 10 | 106–7 | 325–450 | 6.5–70 | 92 ± 10 |
| 4 | 422–439 | 8–96 | 668 ± 50 | 1.5 | 422–439 | 8–96 | 690 ± 50 | – | 422–439 | 8–96 | 660 ± 50 | |
| 5 | 443–464 | 8–99 | 764 ± 50 | 1.2 | 443–464 | 8.6–99 | 830 ± 50 | – | 443–464 | 8.6–99 | 650 ± 50 | |
| 6 | 470–495 | 2–24 | 593 ± 50 | 4.0 | 470–495 | 2–24 | 425 ± 10 | – | 470–495 | 2–24 | 670 ± 50 | |
| 495–544 | 24–99 | 300 ± 50 | 0.9 | 495–544 | 24–99 | 290 ± 10 | 1017–19 | 495–544 | 24–99 | 340 ± 50 | ||
| ДСТ + 10%ПЛА | 2 | 230–285 | 2–47 | 90 ± 10 | 0.9 | 230–285 | 0.5–47 | 94 ± 10 | 106 | 230–285 | 2–47 | 109 ± 10 |
| 285–315 | 47–95 | 162 ± 10 | 1.5 | 285–315 | 47–95 | 162 ± 10 | 1012–13 | 285–315 | 47–95 | 160 ± 10 | ||
| 3 | 330–365 | 4–45 | 102 ± 10 | 1.0 | 325–370 | 3.5–52 | 102 ± 10 | 106–7 | 325–370 | 3.5–52 | 140 ± 10 | |
| 365–400 | 45–92 | 307 ± 10 | 0.9 | 375–395 | 52–93 | 317 ± 50 | 1023 | 370–395 | 52–93 | 217 ± 50 | ||
| 4 | 405–420 | 7–40 | 210 ± 10 | 1.0 | 405–420 | 7–39 | 214 ± 50 | 1016 | 405–420 | 7–40 | 205 ± 50 | |
| 420–425 | 40–99 | 610 ± 50 | 2.9 | 420–430 | 39–99 | 882 ± 50 | – | 420–430 | 40–99 | 760 ± 50 | ||
| 5 | 425–450 | 1–35 | 252 ± 50 | 0.75 | 430–445 | 2–31 | 208 ± 50 | 1016 | 430–445 | 2–30 | 228 ± 50 | |
| 722 ± 50 | ||||||||||||
| 450–460 | 35–91 | 760 ± 50 | 2.0 | 445–460 | 31–91 | 286 ± 10 | – | 445–470 | 30–98 | 740 ± 50 | ||
| 6 | 485–500 | 10–50 | 317 ± 50 | 1.25 | 485–525 | 10–80 | 117 ± 10 | 1020–21 | 485–525 | 10–80 | 320 ± 50 | |
| 510–550 | 50–99 | 152 ± 50 | 0.7 | 525–550 | 80–99 | 162 ± 10 | 108–9 | 525–550 | 80–99 | 120 ± 10 | ||
| ДСТ +1 0% ПЛА + 2.5% ТМДЭТА | 1 | 150–175 | 3–90 | 190 ± 50 | 3–90 | 150–175 | 3–90 | 196 ± 50 | 1022 | 150–175 | 3–90 | 212 ± 5 |
| 2 | 235–320 | 3–95 | 126 ± 10 | 1.0 | 235–320 | 3–91 | 126 ± 10 | 1010–11 | 235–315 | 3–91 | 126.5 ± 10 | |
| 3 | 325–370 | 1–53 | 107 ± 10 | 1.0 | 325–370 | 1–53 | 115 ± 10 | 107 | 325–365 | 1–50 | 149 ± 10 | |
| 370–395 | 53–94 | 314 ± 10 | 1.2 | 370–395 | 53–93 | 250 ± 10 | 1017–18 | 365–395 | 50–94 | 260 ± 10 | ||
| 4 | 410–430 | 7.8–74 | 610 ± 50 | 1.0 | 410–430 | 1–99 | 610 ± 50 | – | 405–435 | 0.4–99 | 590 ± 30 | |
| 5 | 430–455 | 1–78 | 860 ± 50 | 0.9 | 430–455 | 1–80 | 850 ± 50 | – | 430–455 | 1–78 | 830 ± 50 | |
| 455–470 | 78–99 | 550 ± 50 | 1.0 | 455–470 | 80–99 | 520 ± 50 | – | 455–470 | 78–99 | 372 ± 50 | ||
| 6 | 480–510 | 5–53 | 300 ± 50 | 1.25 | 480–510 | 5–53 | 306 ± 10 | 1011–12 | 470–500 | 3–38 | 345 ± 10 | |
| 510–545 | 53–86 | 180 ± 10 | 0.7 | 510–545 | 53–86 | 190 ± 10 | 1010 | 500–525 | 38–76 | 184 ± 10 | ||
| 545–580 | 86–99 | 92 ± 10 | 0.4 | 545–580 | 86–99 | 106 ± 10 | 106 | 525–585 | 76–99 | 103.5 ± 10 | ||
| ДСТ + 10% ПЛА + 2.5% ДСДМАХ | 2 | 235–300 | 0.4–97 | 57 ± 10 | 1.0 | 235–300 | 8.6–99 | 46 ± 10 | 101–2 | 235–305 | 3–97 | 64 ± 10 |
| 3 | 305–370 | 8.6–97 | 121 ± 10 | 1.0 | 305–370 | 8.6–97 | 117 ± 10 | 108 | 305–370 | 8.6–97 | 92 ± 10 | |
| 4 | 360–410 | 8.6 –94 | 154 ± 10 | 0.9 | 360–410 | 5–94 | 163 ± 10 | 1011 | 365–415 | 8.6–99 | 214 ± 10 | |
| 5 | 415–445 | 8.6–97 | 312 ± 50 | 1.2 | 410–455 | 8.6–97 | 468 ± 10 | – | 410–445 | 8.9–97 | 560 ± 10 | |
| 6 | 480–560 | 5–94 | 154 ± 10 | 0.96 | 480–560 | 5–94 | 186 ± 10 | 1011 | 480–560 | 5–94 | 189 ± 10 | |
В качестве примера на рис. 5 представлены кривые в координатах модели Friedman–Ozawa для ПЛА и композиций ДСТ + 10% ПЛА, ДСТ + 10% ПЛА + 2.5% ДСДМАХ в интервале температур, характерных для пиков 2 и 3. Этот рисунок отчетливо демонстрирует существование различий между наклонами кривых, следовательно, между величинами Ea, в температурных областях, принадлежащих разным пикам, а также одному и тому же пику 2 как у индивидуального ПЛА, так и у композиций. Таким образом, введение наполнителей в ДСТ приводит к изменению механизма деструкции его компонентов.
Рис. 5.
Полулогарифмические температурные зависимости скоростей превращения полимеров в ходе деструкции, представленные моделью Friedman–Ozawa: 1 − интервал температур (230–400°С), характерный для пика 2 ПЛА; 2 − композиция ДСТ + 10% ПЛА; 3 − суммарная зависимость для пиков 2 и 3 композиции ДСТ + 10% ПЛА + 2.5% ДСДМАХ.
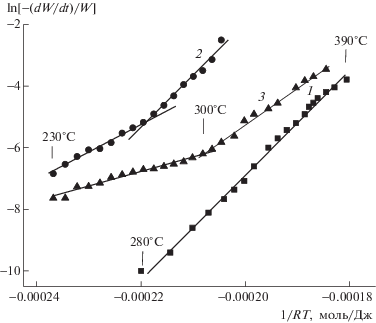
Кроме того, на рис. 5 показано как изменяется механизм распада полимера как при переходе от температурной области одного пика к другому, например от второго к третьему у системы ДСТ–ПЛА–ДСДМАХ, так и внутри одного пика 2 у ДСТ–ПЛА. Из анализа данных табл. 2 следует, что при n = 1.2 величины Еа разложения цепей ПЛА, определенные методами Freeman–Carroll, Friedman–Ozawa и Coats–Redfern составляет 138 ± 10, 144 ± 10 и 154 ± 10 кДж/моль соответственно (табл. 2), при этом значение предэкспоненты A равно 1010–11. В дальнейшем обсуждаемые в работе данные будут относиться к величинам, определенным из соотношений Friedman–Ozawa.
Указанная величина энергии активации отвечает процессу отрыва связей >C=0 [39–41]. При деструкции ПЛА протекает гидролиз, цепи разрушаются по связи С(О)–О и сшиваются в результате отрыва протона от группы с Ea = 105 ± ± 10 кДж/моль [42–45].
На линеаризованной зависимости Friedman–Ozawa для пиков 2 и 3 у образцов ДСТ и композиции ДСТ–ПЛА (рис. 5) обнаруживаются перегибы, свидетельствующие о двустадийности процесса распада компонента ПЛА в матрице ДСТ. По-видимому, на начальной стадии деструкции разрываются слабые связи С(О)–О фазы ПЛА. Такие связи могут возникать в результате межмолекулярных взаимодействий ПЛА с цепями полибутадиена, содержащего преимущественно структуру 1,2-ПБ, у границ ПС-блоков макромолекул ДСТ. Следует отметить, что взаимному проникновению ПЛА и 1,2-ПБ способствует рыхлая структура каучуковой фазы в ДСТ. При более глубокой стадии процесса рост Еа обусловлен деструкцией молекул ПЛА, взаимодействующих с ПБ. Это взаимодействие приводит к возникновению стерических препятствий, снижению сегментарной подвижности ПЛА и торможению распада его функциональных связей. При этом начинают распадаться более прочные углерод-углеродные связи ПБ, на что указывают изменения как величин эффективных энергий активации, так и предэкспонент (табл. 2). Потеря массы композиции составляет 4.25 и 9.75% при 285 и 325°С соответственно, в то время как потеря массы ДСТ при тех же температурах ниже и равна 1.25 и 2.5%. Таким образом, введение ПЛА в ДСТ не только ускоряет его деструкцию, но и изменяет механизм процесса.
Изменение в механизме деструкции ДСТ можно установить при сравнении процесса разрушения цепей в структуре 1,2-ПБ, характеризующегося пиком 3 в интервале температур 325–420°С. В реакции деструкции 1,2-ПБ, при которой расщепляются слабые связи С–С в цепи с образованием смеси олигомеров, энергия активации составляет 125–140 кДж/моль [45]. В настоящем эксперименте разрушение от 6.5 до 70% ПБ цепей в чистом ДСТ протекает с Еа = 112 ± 10 кДж/моль (табл. 2). Введение ПЛА в ДСТ приводит к двустадийному деструкционному процессу в температурных интервалах 325–370 и 375–395°С с величинами Еа1 и Ea2 равными 102 ± 10 и 317 ± 10 кДж/моль соответственно (табл. 2). При этом степень превращения ПБ в данных температурных интервалах изменяется в пределах 3.5–53 и 53–94.
Если термодеструкция протекает через промежуточные радикалы с передачей кинетической цепи, ее энергия активации составляет от ~92 до 120 кДж/моль [45]. По-видимому, некоторое снижение энергии активации разрушения макромолекул 1.2-ПБ (до Ea1) в интервале 285–370°С инициировано распадом слабых связей ПЛА. Увеличение Ea2 может быть обусловлено распадом связей С–С главных цепей 1,2-ПБ и образованием олигомеров разной длины. Этот процесс может сопровождаться передачей цепи как путем внутримолекулярной реакции переноса радикалов ПБ, так с участием радикалов ПЛА и, возможно, связей ПС, локализованных на стыке между ПБ- и ПС-блоками.
В системе ДСТ/ПЛА с ТМДЭТА в интервале температур, характерных для пика 2 (235–320°С), на полулогарифмической зависимости Friedman–Ozawa проявляется один участок с Еа = 126 ± ± 10 кДж/моль, соответствующий степени преврашения ПЛА 3.4−90% (табл. 2). По видимому, в данной системе эффективная энергия активации термодеструкции ПЛА имеет значение промежуточное между теми, которые характеризуют разрушение слабых связей С(О)–О и более прочных связей С–С для композиции ДСТ–ПЛА. В температурном интервале пика 3 термодеструкция системы ДСТ–ПЛА–ТМДЭТА двустадийна, аналогично ДСТ–ПЛА. Но в отличие от последней на втором температурном интервале 370−395°С у системы с ТМДЭТА величины энергии активации и предэкспоненты существенно падают (табл. 2), что свидетельствует об увеличении скорости разрушения связей 1,2-ПБ.
Особенности разрушения системы ДСТ–ПЛА–ТМДЭТА могут быть связаны с влиянием ТМДЭТА на межмолекулярные взаимодействия фазы ПЛА с 1,2-ПБ в ДСТ, приводящие к преобладанию процесса отрыва концевых групп в ПЛА.
Добавление ДСДМАХ в смесь ДСТ/ПЛА приводит к более однородному разрушению цепей в интервале 360–415°С. Процесс протекает с энергией активации 163 ± 10 кДж/моль и A = 1011, в конце этого интервала (к 415°С) степень превращения цепей ПБ достигает 99%, а потеря массы образца 32.5%.
Важно отметить, что в температурном интервале ~440–460°С, характеризуемом пиками 4 и 5 обнаруживаются аномально высокие значения энергий активации. Например, у индивидуального ДСТ в указанной области температур для пиков 4 и 5 величина Еа составляет 690 ± 50 и 830 ± ± 50 кДж/моль. Согласно литературным данным [45], такие значения Ea обусловлены активным распадом цепей в блоках ПБ, имеющих структуру цис-1,4-ПБ и транс-1,4-ПБ с образованием летучих продуктов. Как следует из рис. 2 и табл. 1, наибольшая скорость разрушения полимера и основная часть общей потери массы образцов приходится именно на эти температурные области. Высокие значения Еа могут быть результатом наложения эндотермы распада связей С–С с образованием летучих продуктов на эндотерму их испарения. Данный эффект характерен для структур 1,4-цис и 1,4-транс ПБ и проявляется в интервале 443–464°С [45].
Введение ПЛА в ДСТ резко увеличивает скорость распада и энергию активации процесса разрушения данных структур. В то же время введение ТМДЭТА и ДСДМАХ значительно их уменьшает. Стадии распада в области пиков 4, 5 протекают с меньшей максимальной скоростью (табл. 1) и имеют меньшие значения Еа (табл. 2). Увеличение указанных параметров в образцах ДСТ–ПЛА и уменьшение в системах с ТМДЭТА и ДСДМАХ, очевидно обусловлены инициирующим действием на распад 1.4-ПБ в случае первого из этих ПАВ и тормозящим действием – второго.
Необходимо отметить особенность разрушения ПС-блоков в ДСТ в индивидуальном полимере и в его композициях. У образца исходного ДСТ (пик 6), ПС-блоки имеют плотную упаковку. Если термическое разрушение гомо-ПС идет в одну стадию, а основным продуктом является стирол, то в блоках ПС в структуре ДСТ их деструкция протекает с расщеплением цепи на более короткие фрагменты с образованием мономеров и смеси олигомеров разной длины и строения. В связи с этим наблюдаются две стадии разрушения ПС цепей причем на второй стадии величины Еа и А значительно понижаются (табл. 2). Уменьшение кинетических параметров разрушения ПС-блоков в системе ДСТ–ПЛА вызывает распад макромолекул ПЛА с образованием макрорадикалов, которые локализованы в фазе ПБ на границе с ПС-блоками и инициируют разрыв связей в этих блоках.
Введение ТМДЭТА в систему ДСТ–ПЛА увеличивает гетерогенность структуры ПС-блоков, о чем свидетельствует уширение пика 6 и уменьшение энергии активации их деструкции (табл. 2). Можно выделить три стадии, предположительно различающиеся механизмом процесса. Эти стадии (по Friedman–Ozawa) в пределах температурных интервалов 480–510, 510–545, 545–580°С соответствуют степеням превращения ПС-цепей 5–53, 53–86, 86–99% и характеризуются величинами Еа = 306 ± 10, 190 ± 10, 106 ± 10 кДж/моль (табл. 2). Более высокие значения величины энергии активации в этом ряду, по-видимому, свидетельствуют о деструкции связей С–С в блоках ПС, цепи которых характеризуются высокой плотностью и низкой сегментарной подвижностью. Низкие величины Еa, возможно, связаны с деструкцией, протекающей с участием радикалов.
Введение ДСДМАХ в систему ДСТ–ПЛА приводит к гомогенезации процесса деструкции блоков ПС (пик 6) и в результате – к существенному снижению значений кинетических параметров. В указанной системе деструкция блоков ПС протекает в интервале температур 480−560°С до степени превращения звеньев 94% по одному механизму с Еа = 186 ± 10 кДж/моль (табл. 2). Данная величина энергии активации может указывать на гомолитический разрыв связей С–С в блочных структурах ПС. При этом локализованный в них ДСДМАХ тормозит процесс, выполняя роль ингибитора деструкции.
Таким образом, из полученных данных следует, что биодеградируемый полимер ПЛА и поверхностно активные добавки, введенные в ДСТ, оказывают различное влияние на кинетику и механизм термодеструкции компонентов ДСТ вследствие различий в природе наполнителей и в их локализации.
Разложение композиций, содержащих ПЛА, увеличивает, а композиций, содержащих ПАВ, снижает потерю массы ДСТ в результате уменьшения выхода газообразных продуктов из зоны деструкции полимерной матрицы. Основным фактором, определяющим характер изменения кинетики термодеструкции, можно назвать наличие межмолекулярных взаимодействий добавок ПЛА и ПАВ с эластомерной и термопластичной компонентами в полимерной матрице из ДСТ. Характер этого взаимодействия обусловлен локализацией полилактида в бутадиеновых блоках бутадиен-стирольной матрицы вокруг доменов полистирола, а поверхностно-активных веществ – в полилактиде и матрице бутадиен-стирольного термоэластопласта, включая блоки ПС. Жесткость блоков ПС препятствует проникновению в них ПЛА.
Авторы статьи выражают благодарность А.А. Ильину за его помощь в экспериментальной работе и в обсуждении результатов.
Список литературы
Полимерные пленки / Под ред. Е.М. Абдель-Бари. СПб.: Профессия, 2005.
Gomzyak V.I., Sedush N.G., Puchkov A.A., Polyakov D.K., Chvalun S.N. // Polymer Science B. 2021. V. 63. № 3. P. 257.
Krasavtsev V., Maslova G., Noudga L., Petrova V., Ezhova E., Krivosheina L. // Adv. Chitin Science, Montreal, Canada. 2003. V. 7. P. 269.
Лекишвили М.В., Панасюк А.Ф. // Вестн. РАМН. 2008. № 9. С. 33.
Misra S., Ansari T., Valappil S. // Biomaterials. 2010. № 31. P. 2806.
Artsis M.I., Bonartsev A.P., Iordanskii A.L., Bonartseva G.A., Zaikov G.E. // Molec. Cryst. Liq. Cryst. 2012. V. 555. P. 232.
Paşcu E.I., Stokes J., McGuinness G.B. // Mater. Sci. Eng. A. 2013. № 33. P. 4905.
Shumilova A.A., Nikolaevaa E.D. // J. Siberian Federal Univ. Biology. 2016. V. 1. № 9. P. 53.
Sombatmankhong K., Suwantong O., Waleetorncheepsawat S., Supaphol P. //J. Polym. Sci., Polym. Phys. 2006. V. 44. № 19. P. 2923.
Shibryaeva L.S., Makarov O.V., Andryukhin M.I., Lyusova L.R., Il’in A.A. // Polymer Science D. 2015. T. 8. № 1. P. 75.
Borovkova N.V., Evseev A.K., Makarov M.S., Goroncharovskaya Ir.V., Vinogradova O.N., Nikolaeva E.D., Goncharov D.B. // J. Siberian Federal Univ. Biology. 2016. V. 9. № 1. P. 43.
Volova T.G., Vinogradova O.N., Zhila N.O., Kiselev E.G., Peterson I.V., Vasil’ev A.D., Sukovatyi A.G., Shishatskaya E.I. // Polymer Science A. 2017. V. 59. № 1. P. 98.
Жуковский В.А. // Научный электронный журнал “NNOVA”. 2016. № 2 (3). С. 1.
Grassie N., Scott G. Polymer Degradation and Stabilisation Cambridge: Cambridge Univ. Press, 1985.
Каминский В.А., Кузнецов А.А. // Теорет. основы хим. технологии. 2012. Т. 46. № 4. С. 453.
Крутько Э.Т., Прокопчук Н.Р., Глоба А.И. Технология биоразлагаемых полимерных материалов Уч. метод. пособие. Минск: Белорусский гос. технол. ун-т, 2014.
Shibryaeva L.S., Krasheninnikov V.G., Gorshenev V.N. // Polymer Science A. 2019. V. 61. № 2. P. 162.
Люсова Л.Р., Ильин А.А., Ковалева А.Н., Шибряева Л.С., Карпова С.Г., Макаров О.В., Тарасюк В.Т., Грачёва А.Ю. Состав эластичного антибактериального материала. Пат. 2629603. Россия // Б.И. 2017. № 25. С. 7.
Shibryaeva L.S., Makarov O.V., Andryukhin M.I., Lyusova L.R., Il’in A.A. // Polymer Science D. 2015. V. 8. № 1. P. 75.
Фойгт И. Стабилизация синтетических полимеров против действия света и тепла. Пер. с нем. Л.: Химия, 1972.
Термоэластопласты / Под ред. В.В. Моисеева. М.: Химия, 1985..
Мурзаканова М.М., Залова Т.В., Борукаев Т.А., Микитаев А.К. // Пласт. массы. 2010. № 8. С. 3.
Polymer Composite Materials: Structure, Properties, Technology / Ed. By A.A. Berlin. CPb: Professiya, 2014.
Shibryaeva L.S., Reshmin Yu.A., Kuksenko E.S., Shatalova O.V., Krivandin A.V., Gorbunova I.Yu., Kerber M.L. // Polymer Science A. 2007. V. 49. № 1. P. 12.
Кулезнев В.Н. Смеси и сплавы полимеров. СПб.: Научные основы и технологии, 2013.
Прокопчук И.Р. // Пласт. массы. 1983. № 10. С. 24.
Menczel J.D., Prime R.B. Thermal Analysis of Polymers, Fundamentals and Applications. New York: Wiley, 2009.
Chan J.H., Balke S.T. // Polym. Degrad. Stab. 1997. V. 57. № 1. P. 135.
Flammersheim H.-J., Opfermann J.R. // Macromol. Mat. Eng. 2001. V. 286. № 3. P. 143.
Herman M.F. Encyclopedia of Polymer Science and Technology. New York: Wiley., 2005. V. 2.
Starink M.J. // Thermochim. Acta. 2003. V. 404. № 1. P. 163.
Ozawa T.J. // J. Thermal Analysis. 1986. V. 31. № 3. P. 547.
Ozawa T.J. // Thermochim Acta. 2000. V. 355. № 1–2. P. 35.
Ozawa T.J. // J. Therm. Anal. Calorim. 2005. V. 82. № 3. P. 687.
Ginzburg B.M. // Polymer Science A. 2012. V. 54. № 3. P. 248.
Moukhina E. // J. Therm. Anal Calorim. 2012. V. 109. № 3. P. 1203.
Moukhina E. 39 North American Thermal Analysis Society (NATAS) Conference. Des Moines Lowa, 2011. V. 1.
Пен В.Р. // Химия растительного сырья. 2004. № 2. С. 101.
Сумин М.И. Методы регуляции А.Н. Тихонова для решения оперативных уравнений первого рода. Уч.-методич. пособие. Нижний Новгород: Нижегородский гос. ун-т, 2016.
Kratkiy spravochnik phizico-khimicheckih velichin/ Ed. by K.P. Mishchenko, A.A. Ravdel`. Leningrad: Khimiya, 1974.
Arshady R. // J. Controlled Release. 1991. V. 17. P. 1.
Juni K., Nakano M., Kubota M. // J. Controlled Release. 1986. V. 4. P. 25.
Uhrich K.E., Cannizzaro S.M., Langer R.S., Shakesheff K.M. // Chem. Rew. 1999. V. 99. P. 3181.
Gunatillake P.A., Adhikari R. // Eur. Cells Materials. 2003. V. 5. P. 1.
Справочник химика. Л.; М.: Госхимиздат, 1951–1952.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)